- Discovery and development of HIV-protease inhibitors
-
Many major physiological processes depend on regulation of proteolytic enzyme activity and there can be dramatic consequences when equilibrium between an enzyme and its substrates is disturbed. In this prospective, the discovery of small-molecule ligands, like protease inhibitors, that can modulate catalytic activities has an enormous therapeutic effect.[1] Hence, inhibition of the HIV protease is one of the most important approaches for the therapeutic intervention in HIV infection[2] and their development is regarded as major success of structure-based drug design.[3] They are highly effective against HIV[4] and have, since the 1990s, been a key component of anti-retroviral therapies for HIV/AIDS.[5]
Contents
History
Human immunodeficiency virus (HIV) is a lentivirus that has two major species, HIV-1 which causes the majority of the epidemic, and HIV-2, a close relative whose distribution is concentrated in western Africa.[6] HIV infection was first described in 1981 in San Francisco and New York City.[7] In 1985, HIV was identified as the causative agent of acquired immune deficiency syndrome (AIDS) and its complete genome was immediately available. This knowledge paved the way for the development of selective inhibitors.[6] HIV-2 carries a slightly lower risk of transmission than HIV-1 and infection tends to progress more slowly to AIDS.[7] In common usage HIV usually implies HIV-1.[8] HIV-1 protease is one of the best known aspartic proteases, and an attractive target for the treatment of AIDS.[9] After the discovery of HIV protease it only took 10 years for its first inhibitor to reach the market.[10] The first reports of highly selective antagonists against the HIV protease were revealed in 1987. Phase I trials of saquinavir began in 1989 and it was the first HIV protease inhibitor to be approved for prescription use in 1995. Four months later, two other protease inhibitors, ritonavir and indinavir, were approved.[6] In 2009, ten protease inhibitors have reached the market for treatment against HIV but one protease inhibitor, amprenavir, was withdrawn from the market in 2004.[6][11]
Life cycle of HIV
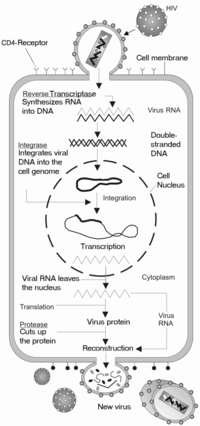 The HIV replication cycle
The HIV replication cycle
HIV belongs to the class of viruses called retroviruses, which carry genetic information in the form of RNA. HIV infects T cells that carry the CD4 antigen on their surface. When HIV infects its target cell it requires fusion of the viral and cellular membranes.[12] The first step is the interaction between envelope proteins of the virus (gp120, gp41) and specific host-cell surface receptors (e.g. CD4 receptor) on the target cell. Then the virus binds to the chemokine coreceptors CXCR4 or CCR5, resulting in conformational changes in the envelope proteins. This fusion creates a pore through which the viral capsid enters the cell.[13] Following entry into the cell the RNA of the virus is reverse-transcribed to DNA by the first virally encoded enzyme, the reverse transcriptase. The viral DNA enters the nucleus where it is integrated into the genetic material of the cell by the integrase, a second virally encoded enzyme. Activation of the host cell leads to the transcription of the viral DNA into mRNA. The mRNA is then translated into viral proteins and the third virally encoded enzyme, namely HIV protease, is required to cleave a viral polyprotein precursor into individual mature proteins. The viral RNA and viral proteins assemble at the surface of the cell into new virions. The virions bud from the cell and are released to infect other cells. All infected cells are eventually killed because of this extensive cell damage, from the destruction of the host's genetic system to the budding and release of virions.[12]
Mechanism of action
There are several steps in the HIV life cycle that may be interfered with, thus stopping the replication of the virus. A very critical step is the proteolytic cleavage of the polypeptide precursors into mature enzymes and structural proteins catalyzed by HIV protease.[12] HIV protease inhibitors are peptide-like chemicals that competitively inhibit the action of the virus aspartyl protease. These drugs prevent proteolytic cleavage of HIV Gag and Pol polyproteins that include essential structural and enzymatic components of the virus. This prevents the conversion of HIV virus particles into their mature infectious form.[6] Protease inhibitors can alter adipocyte metabolism causing lipodystrophy, a common side effect associated with the use of most HIV protease inhibitors. Many mechanisms have been proposed, for example inhibition of adipocyte differentiation, triglyceride accumulation and increased lipolysis. Theories considering the effect of protease inhibitors on insulin-stimulated glucose uptake have also been linked to the lipodystrophic syndrome. It is possible that protease inhibitors can cause a decrease in insulin-stimulated tyrosine phosphorylation of IRS-1, representing inhibition of early steps in insulin signaling. Decreased adiponectin secretion and induced expression of interleukin-6 associated with HIV protease inhibitors may also contribute to inhibition of insulin-stimulated glucose uptake.[14]
Design
Protease inhibitors were designed to mimic the transition state of the protease's actual substrates. A peptide linkage consisting of –NH-CO- is replaced by an hydroxyethylen group (-CH2-CH(OH)-) which the protease is unable to cleave. HIV protease inhibitors fit the active site of the HIV aspartic protease and were rationally designed utilizing knowledge of the aspartyl protease's mode of action. The most promising transition state mimic was hydroxyethylamine which lead to the discovery of the first protease inhibitor, saquinavir. Following that discovery, other HIV protease inhibitors were designed using the same principle.[15]
Binding site
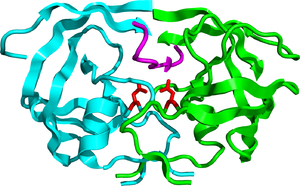 A schematic structure of a HIV-1 protease. The monomers are shown in green and cyan, the Asp-25 and Asp-25´ residues are shown in red, and Ile50 and Ile50´ residues linked to a water molecule are shown in purple.
A schematic structure of a HIV-1 protease. The monomers are shown in green and cyan, the Asp-25 and Asp-25´ residues are shown in red, and Ile50 and Ile50´ residues linked to a water molecule are shown in purple.The HIV protease is a C2-symmetric homodimeric enzyme consisting of two 99 amino acid monomers. Each monomer contributes an aspartic acid residue that is essential for catalysis,[6] Asp-25 and Asp-25´. The HIV protease has the sequence Asp-Thr-Gly, which is conserved among other mammalian aspartic protease enzymes. An extended beta-sheet region on the monomers, known as the flap, constitutes in part the substrate binding site with the two aspartyl residues lying on the bottom of a hydrophobic cavity.[12][16][17] Each flexible flap contains three characteristic regions: side chains that extend outward (Met46, Phe53), hydrophobic chains extending inward (Ile47, Ile54), and a glycine rich region (Gly48, 49, 51, 52). Ile50 remains at the tip of the turn and when the enzyme is unliganded a water molecule makes hydrogen bonds to the backbone of Ile50 on each monomer.[17]
HIV proteases catalyze the hydrolysis of peptide bonds with high sequence selectivity and catalytic proficiency. The mechanism of the HIV protease shares many features with the rest of the aspartic protease family although the full detailed mechanism of this enzyme is not fully understood.[12] The water molecule seems to play a role in the opening and closing of the flaps as well as increasing the affinity between enzyme and substrate. The aspartyl residues are involved in the hydrolysis of the peptide bonds.[17] The preferred cleavage site for this enzyme is the N-terminal side of proline residues, especially between phenylalanine and proline or tyrosine and proline.[6][16]
Development
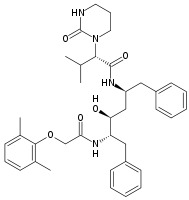
The first HIV protease inhibitor, saquinavir, is a peptidomimetic hydroxyethylamine[6] and was marketed in 1995.[18] It is a transition state analogue of a native substrate of the protease.[6] The observation that HIV-1 protease cleaves the sequences containing the dipeptides Tyr-Pro or Phe-Pro was the basic design criterion.[19] Addition of the decahydroisoquinoline (DIQ) group was one of the most significant modifications that led to the discovery of saquinavir. This substituent improves aqueous solubility and potency by limiting the conformational freedom of the inhibitor.[20] Saquinavir is effective against both HIV-1 and HIV-2[5] and is usually well tolerated but high serum concentration is not achieved.[11]
Ritonavir, a peptidomimetic HIV protease inhibitor, was marketed in 1996.[18] It was designed to fit the C2-symmetry in the binding site of the protease.[6] The developers of ritonavir, Abbott Laboratories, started with compounds that were active against the virus but had poor bioavailability. Some improvements were made, for example the terminal phenyl residues were removed and pyridyl groups put instead to add water solubility. The final product of these improvements was ritonavir.[19] Significant gastrointestinal side effects and a large pill burden are ritonavir's main drawbacks and is therefore not used as a single treatment.[11] However, it is a strong inhibitor of the cytochrome P450 enzyme mediated metabolism[19] and it is only used in a combination therapy with other protease inhibitors for pharmacokinetic boosting.[11]
Indinavir, which is a peptidomimetic hydroxyethylene HIV protease inhibitor, reached the market in 1996.[6][18] The design of indinavir was guided by molecular modeling and the X-ray crystal structure of the inhibited enzyme complex. The terminal phenyl constituents contribute hydrophobic binding to increase potency.[19] It is an analogue of the phenylalanine-proline cleavage site of the HIV Gag-polyprotein.[6]
Nelfinavir was the first protease inhibitor that was not peptidomimetic. In the design process of nelfinavir, an orally bioavailable and nonpeptidic inhibitor, iterative protein cocrystal structure analysis of peptidic inhibitors was used and parts of the inhibitors were replaced by nonpeptidic substituents.[19] Nelfinavir contains a novel 2-methyl-3-hydroxybenzamide group, whereas its carboxyl terminal contains the same DIQ group as saquinavir.[19] Nelfinavir was marketed in 1997[18] and was the first protease inhibitor to be indicated for pediatric AIDS.[19]
Amprenavir reached the market in 1999.[18] It is an N,N-disubstituded amino-sulfonamide nonpeptide HIV protease inhibitor[6] and shares some common features with previous protease inhibitors. It has a core similar to that of saquinavir but with different functional groups on both ends. On one end it has a tetrahydrofuran carbamate group and on the other end is an isobutylphenyl sulfonamide with an added amide. This structure results in fewer chiral centers, that makes it easier to synthesize and gives it enhanced aqueus solubility. That in turn gives better oral bioavailability.[19] However, amprenavir was withdrawn from the market in 2004 since fosamprenavir, its prodrug, proved superior in many aspects.[6]
Lopinavir was marketed in 2000[18] and was originally designed to diminish the interactions of the inhibitor with Val82 of the HIV-1 protease, a residue that is often mutated in the drug resistant strains of the virus.[19] It is a peptidomimetic HIV protease inhbitor[6] and its core is identical to that of ritonavir. Instead of the 5-thiazolyl end group in ritonavir, lopinavir has a phenoxyacetyl group and the 2-isopropylthiazolyl group in ritonavir was replaced by a modified valine in which the amino terminal had a six-membered cyclic urea attached.[19]
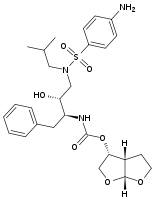
Fosamprenavir was marketed in 2003[18] and is a phosphoester prodrug that is rapidly and extensively metabolized to amprenavir.[21] The solubility and bioavailability is better than of amprenavir[6] which results in reduced daily pill burden.[22]
Atazanavir was marketed in 2003[18] and is an azapeptide protease inhibitor[18] designed to fit the C2-symmetry of the enzyme binding site.[11] Atazanavir showed better resistant profiles than previous HIV protease inhibitors.[4] It is unique among the other protease inhibitors as it can only be absorbed in an acidic environment.[11]
Tipranavir is a nonpeptidic HIV-1 protease inhibitor[11] and reached the market in 2005.[18] Unlike other HIV protease inhibitors on the market, tipranavir was developed from a nonpeptidic coumarin template and its antiprotease activity was discovered by high-throughput screening.[23] This sulfonamide containing 5,6-dihydro-4-hydroxy-2-pyrone had emerged from screenings of 3-substituted coumarins and dihydropyrones.[24] It possesses broad antiviral activity against multiple protease inhibitor resistant HIV-1.[25]
Darunavir reached the market in 2006[18] and is a nonpeptidic analogue of amprenavir, with a critical change at the terminal tetrahydrofuran (THF) group. Instead of a single THF group, darunavir contains two THF groups fused in the compound, to form a bis-THF moiety which makes it more effective than amprenavir. With this structural change, the stereochemistry around the bis-THF moiety confers orientational changes, that allows for continued binding with the protease which has developed a resistance for amprenavir.[26]
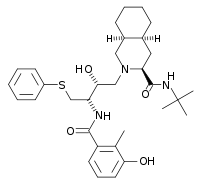
All the FDA approved protease inhibitors are listed below.HIV protease inhibitors the FDA has approved 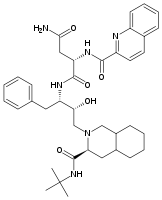
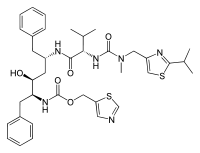
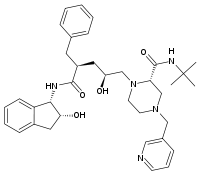
Saquinavir Ritonavir Indinavir Nelfinavir 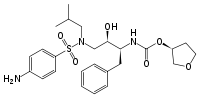
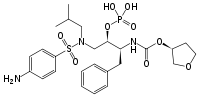
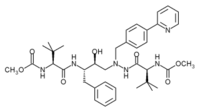
Amprenavir Lopinavir Fosamprenavir Atazanavir 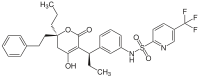
Tipranavir Darunavir Structure-activity relationship
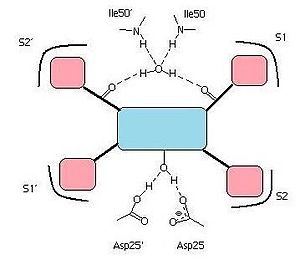 A simplified image of a protease inhibitor binding to the active site of the HIV-1 protease. The central core motif is shown in blue with the hydroxyl group forming hydrogen bonds with Asp-25 and Asp-25´. Hydrogen bonds also connect carbonyl groups on the inhibitor to the water molecule linked to Ile50 and Ile50´. Hydrophobic groups are shown in pink and their complementing pockets referred to as S1, S1´, S2 and S2´.
A simplified image of a protease inhibitor binding to the active site of the HIV-1 protease. The central core motif is shown in blue with the hydroxyl group forming hydrogen bonds with Asp-25 and Asp-25´. Hydrogen bonds also connect carbonyl groups on the inhibitor to the water molecule linked to Ile50 and Ile50´. Hydrophobic groups are shown in pink and their complementing pockets referred to as S1, S1´, S2 and S2´.All the HIV protease inhibitors on the market contain a central core motif consisting of a hydroxyethylen scaffold, with the only exception being the central core of tipranavir, which is based on a coumarin scaffold.[15] A very important group on the HIV protease inhibitors is a hydroxyl group on the core motif which forms a hydrogen bond with the carboxylic acid on the Asp-25 and Asp-25´ residues in the binding site.[16][27] Hydrogen bonds between the water molecule, which is linked to Ile50 and Ile50', and carbonyl groups of the peptidomimetic inhibitors seem to connect them with the flap regions.[19] On the other hand, on the nonpeptidic inhibitors, there is a proton acceptor which replaces the tetracoordinated water molecule and interacts directly with the two Ile50 residues on the flap of the enzyme.[28] Specific pockets in the binding site of the HIV protease, often referred to as S1,S1',S2 and S2', recognize hydrophobic amino acids on natural substrates. The potency of inhibitors bearing hydrophobic groups complementing these areas is therefore increased.[29] Some residues in the enzyme binding site are capable of forming hydrogen bonds with hydrophilic groups on the inhibitor, for example with the THF moieties on amprenavir and darunavir. Since darunavir has a bis-THF moiety, instead of a single THF moiety like on amprenavir, it can form more hydrogen bonds and increase binding energy.[26]
Resistance
Mutations that code for alterations of the conformational shape facilitate resistance of HIV to protease inhibitors.[26] The locations of these mutations are primarily in the active site of the HIV protease enzyme as well as outside of the active site. Active site mutations have been shown to directly change the interactions of the inhibitors with the protease whilst non-active site mutations are considered to affect by other mechanisms, like influencing dimer stability and conformational flexibility.[30] Over 100 single gene point mutations have been described, of which at least 26 are specific to protease inhibitors. Of these, there are about 15 primary or major mutations that are significant enough to change drug activity.[26] Many mutated residues have been found in HIV-1 protease which cause drug resistance, for example Leu33 changes to Ile, Val, or Phe; Val82 to Ala, Phe, Leu, or Thr; Ile84 to Val; and Leu90 to Met.[31] Different mutations affects different protease inhibitors. For instance, mutations at Leu90 evidently affect saquinavir and nelfinavir while indinavir activity is affected by mutations at Met46, Val82, and Ile84, and fosamprenavir is affected when Ile50 changes to Val and at Ile84. A combination of mutations can render high-level drug resistance but single mutations normally do not equate with drug resistance to protease inhibitors.[26] The mutations can be divided into primary mutations and secondary mutations. Primary mutations often have only a small effect on resistance. The chemical structures of most protease inhibitors are quite similar, so it is not surprising that some primary mutations lead simultaneously to resistance to multiple protease inhibitors. Cross-resistance is one of the major problems of protease inhibitor treatment.[32] Additional mutations emerging in the protease during continuous protease inhibitor therapy are commonly referred to as secondary mutations. This can lead to high-level protease inhibitor resistance.[32]
There is no general strategy to tackle the problem of drug resistance. Researches directed towards development of new therapies to cure AIDS are focused on avoiding cross-resistance to drugs that are already on the market.[12]Current status
In November 2009 darunavir was still the most recent HIV protease inhibitor to reach the market.[33] In 2006, GlaxoSmithKline discontinued the phase II clinical development of brecanavir, an investigational protease inhibitor for the treatment of HIV, due to insurmountable issues regarding formulation.[34] In the summer of 2009, GlaxoSmithKline and Concert Pharmaceuticals announced their collaboration to develop and commercialise deuterium-containing medicines. One of them is CTP-518, a protease inhibitor for the treatment of HIV, expected to enter phase I clinical trials in the second half of 2009. CTP-518 is a novel HIV protease inhibitor developed by replacing certain key hydrogen atoms of atazanavir with deuterium. Pre-clinical studies have demonstrated that this modification fully retains the antiviral potency but can evidently slow hepatic metabolism and thereby increase the half life and plasma trough levels. CTP-518, therefore, has the potential to be the first HIV protease inhibitor to eliminate the need to co-dose with a boosting agent, such as ritonavir.[35]
See also
- Antiretroviral drug
- Reverse transcriptase inhibitor
- Integrase inhibitor
- Entry inhibitor
- Discovery and development of non-nucleoside reverse transcriptase inhibitors
References
- ^ Cuccioloni, M. et al. (2009) Natural Occurring Polyphenols as Template for Drug Design. Focus on Serine Proteases. Chemical biology and drug design. 74;1-15.
- ^ Chen, X. et al. (2003) Synthesis and SAR Studies of Potent HIV Protease Inhibitors Containing Novel Dimethylphenoxyl Acetates as P2 Ligands. Bioorganic & Medicinal Chemistry Letters. 13;3657-3660.
- ^ Adachi, M. et al. (2009) Structure of HIV-1 protease in complex with potent inhibitor KNI-272 determined by high-resolution X-ray and neutron crystallography. Proceedings of the national academy of sciences of the United States of America. 12; 4641-4646
- ^ a b Yanchunas, J. et al. (2005) Molecular Basis for Increased Susceptibility of Isolates with Atazanavir Resistance-Conferring Substitution I50L to Other Protease Inhibitors. Antimicrobial Agent and Chemotherapy. 40;3825-3832.
- ^ a b Brower, E.T., et al. Inhibition of HIV-2 protease by HIV-1 protease inhibitors in clinical use. Chemical Biology & Drug Design. 71;298-305.
- ^ a b c d e f g h i j k l m n o p Brunton, L.L., Lazo, J.S. and Parker, K.L. (2006) Goodman and Gilmans´s The Pharmacological Basis of Therapeutics (11th edition). United States of America: McGraw-Hill.
- ^ a b http://emedicine.medscape.com/article/211316-overview, retrieved: October 27th 2009
- ^ Kurup, A., Mekapati, S.B., Garg, R. and Hansch, C. (2003) HIV-1 Protease Inhibitors: A Comparative QSAR Analysis. Current Medicinal Chemistry. 10;1679-1688
- ^ Shi, H., Liu, K., Leong, W.W.Y. and Yao, S.Q. (2009) Expedient solid-phase synthesis of both symmetric and asymmetric diol libraries targeting aspartic proteases. Bioorganic & Medicinal Chemistry Letters. 19;3945-3948.
- ^ Turk, B. (2006) Targeting proteases: successes, failures and future prospects. Nature Reviews Drug Discovery. 5;785-799
- ^ a b c d e f g [1] Graziani, A.L., et al(2009), HIV Protease Inhibitors. Retrieved: October 30th 2009
- ^ a b c d e f Brik, A. and Wong, C.H. (2003) HIV-1 protease: mechanism and drug discovery. Organic & Biomolecular Chemistry. 1(1); 5-14.
- ^ Warnke, D., Barreto, J. and Temesgen, Z. (2007) Antiretroviral drugs. Journal of Clinical Pharmacology. 47(12); 1570-1579.
- ^ Kim, R.J., Wilson, C.G., Wabitsch, M., Lazar, M.A. and Steppan, C.M. (2006) HIV protease inhibitor-specific alterations in human adipocyte differentiation and metabolism. Obesity. 14; 994-1002.
- ^ a b De Clercq, E. (2009) The history of antiretrovirals: key discoveries over the past 25 years. Reviews in Medical Virology. 19; 287-299.
- ^ a b c Mimoto, T., Hattori, N., Takaku, H. et al. (2000) Structure-activity relationship of orally potent tripeptide-based HIV protease inhibitors containing hydroxymethylcarbonyl isostere. Chemical & Pharmaceutical Bulletin. 48(9); 1310-1326.
- ^ a b c Perez, M.A.S., Fernandes, P.A. and Ramos, M.J. (2007) Drug design: New inhibitors for HIV-1 protease based on Nelfinavir as lead. Journal of Molecular Graphics and Modelling. 26; 634-642.
- ^ a b c d e f g h i j k Flexner, C. (2007) HIV drug development: the next 25 years. Nature Reviews Drug Discovery. 6; 959-966.
- ^ a b c d e f g h i j k Wlodawer, A. (2002) Rational approach to AIDS drug design through structural biology. Annual Review of Medicine. 53; 595-614.
- ^ Smith, H.J. and Simons, C. (2005) Enzymes and Their Inhibition: Drug Development (6th edition). United State of America: CRC press
- ^ Chapman, T.M., Plosker, G.L. and Perry, C.M. (2004) Fosamprenavir – A Review of its Use in the Management of Antiretroviral Therapy-naive Patients with HIV Infection. Drugs. 64; 2101-2124.
- ^ a b c d e McCoy, C. (2007) Darunavir: A nonpeptidic antiretroviral protease inhibitor. Clinical Therapeutics. 29(8); 1559-1576.
- ^ Liu, F., Kovalevsky, A.Y., Tie, Y., Ghosh, A.K., Harrison, R.W. and Weber, I.T. (2008) Effect of Flap Mutations on Structure of HIV-protease and Inhibition by Saquinavir and Darunavir. Journal of Molecular Biology. 381(1); 102-115
- ^ Lebon, F. and Ledecq, M. (2000) Approaches to the Design of Effective HIV-1 Protease Inhibitors. Current Medicinal Chemistry. 7; 455-477.
- ^ Blum, A. et al. (2008) Achiral oligoamines as versatile tool for the development of aspartic protease inhibitors. Bioorganic & Medicinal Chemistry. 16; 8574-8586.
- ^ Bihani, S. C., Das, A., Prashar, V., Ferrer, J.-L. and Hosur; M.V. (2009) Resistance mechanism revealed by crystal structures of unliganded nelfinavir-resistant HIV-1 protease non-active site mutants N88D and N88S. Biochemical and Biophysical Research Communications. 389; 295-300.
- ^ Lemke,T.L., Williams, D.A., Roche, V.F. and Zito, S.W. (2008) Foye´s Principles of Medicinal Chemistry ( 6th edition). United States of America: Lippincott williams & Wilkins, a Wolters Kluwer business.
- ^ a b Maarseveen, N.V. and Boucher, C. (2008) Antiretroviral Resistance in Clinical Practice. London: Mediscript Ltd.
- ^ De Clercq, E. (2009) Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. International Journal of Antimicrobial Agents. 33; 307-320.
- ^ [2]GSK and Concert Pharmaceuticals form alliance to develop novel deuterium-modified drugs. Retrieved november 4th. 2009.
- ^ [3] GlaxoSmithKline discontinues clinical development of investigational protease inhibitor brecanavir (640385). Retrieved november 4th. 2009.
Categories:- Drug discovery
- HIV/AIDS












Wikimedia Foundation. 2010.