- Discovery and development of dual serotonin and norepinephrine reuptake inhibitors
-
Discovery and development of dual serotonin and norepinephrine reuptake inhibitorsSerotonin–norepinephrine reuptake inhibitors (SNRIs) are a class of antidepressant drugs used in the treatment of major depressive disorder (MDD). SNRIs are potent inhibitors of serotonin (5-Hydroxytryptamine, 5-HT) and norepinephrine (NE, noradrenalin) reuptake. These neurotransmitters are known to play an important role in mood. The human serotonin transporter (SERT) and norepinephrine transporter (NET) are membrane proteins that are responsible for the reuptake of serotonin and norepinephrine. Balanced dual inhibition of monoamine reuptake can possibly offer advantages over other antidepressants drugs by treating a wider range of symptoms[1]. SNRIs are second-generation agents, such as selective serotonin reuptake inhibitors (SSRIs) and norepinephrine reuptake inhibitors (NRIs). Over the past two decades, second-generation agents have gradually replaced first-generation agents, such as tricyclic antidepressants (TCAs) and monoamine oxidase inhibitors (MAOIs) as the drugs of choice for the treatment of MDD. This is mainly because of their improved tolerability and safety profile[2].
Contents
History
In 1952 iproniazid, a antimycobacterial agent, was discovered to have psychoactive properties while researched as a possible treatment for tuberculosis. Researchers noted that patients given iproniazid became cheerful, more optimistic, and more physically active. Soon after its development, iproninazid and related substances were shown to slow enzymatic breakdown of norepinephrine, serotonin and dopamine via inhibition of mitochondrial enzyme monoamine oxidase. For this reason this class of drugs became known as MAOIs. During this time development of distinctively different antidepressant agents was also researched. Imipramine became the first clinically useful TCA. Imipramine was found to affect numerous neurotransmitter systems and to block reuptake of norepinephrine and serotonin from the synapse, therefore increasing the levels of these neurotransmitters. Use of MAOIs and TCAs gave major advances in treatment of depression but their use was limited by unpleasant side effects and significant safety and toxicity issues [3].
Throughout the 1960s and 1970s the catecholamine hypothesis of emotion and its relation to depression was of wide interest and that the decreased levels of certain neurotransmitters, such as norepinephrine, serotonin, and dopamine might play a role in the pathogenesis of depression. This led to the development of fluoxetine, the first SSRI. The improved safety and tolerability profile of the SSRIs in patients with MDD, compared with TCAs and MAOIs, represented yet another important advance in the treatment of depression[3].
Since the late 1980s SSRIs have dominated the antidepressant drug market. Today there is increased interest in antidepressant drugs with broader mechanisms of action that may offer improvements in efficacy and fewer adverse effects. In 1993 a new drug was introduced to the US market called venlafaxine, a serotonin-norepinephrine reuptake inhibitor [4]. Venlafaxine was the first compound described in a new class of antidepressive substances called phenylethylamines. These substances are unrelated to TCA and other SSRIs. Venlafaxine blocks the neuronal reuptake of serotonin, noradrenaline, and, to a lesser extent, dopamine in the central nervous system. In contrast with several other antidepressant drugs, venlafaxine can induce a rapid onset of action mainly due to a subsequent norepinephrine reuptake inhibition[5]. See timeline in figure 1. Figure 1: Timeline of development of antidepressant agents.
Figure 1: Timeline of development of antidepressant agents.
Overview of SNRIs
Venlafaxine (Efexor®, Effexor®) was the first drug described in the class of SNRIs[5]. Venlafaxine was approved by the Food and Drug Administration (FDA) in the US in 1993 for the management of treatment-resistant depression (TRD). Venlafaxine with extended-release was also introduced to the US market for the treatment of generalized anxiety disorder (GAD) as well as chronic pain syndromes[4].
Sibutramine (Meridia®, Reductil®) is used in the treatment of obesity. Sibutramine was approved by the FDA in 1998 and was then a new class of medication. Sibutramine was then the first drug for the treatment of obesity to be approved in 30 years[6].
Duloxetine (Cymbalta®, Ariclaim®, Xeristar®, Yentreve®) was the second SNRI approved by the FDA in 2004. Duloxetine is used in the treatment of MDD and fibromyalgia[7].
Desvenlafaxine (Pristiq®) is the active metabolite of venlafaxine. Desvenlafaxine is used in the treatment of MDD in adults. Desvenlafaxine was approved by the FDA in 2008 and was then the third approved SNRI[8].
Milnacipran (Savella®, Ixel®, Dalcipran®, Toledomin®) was approved by the FDA in 2009 for the treatment of pain and multiple symptoms associated with fibromyalgia syndrome[9]. Milnacipran is also used in the treatment of MDD[10]. See timeline of approved SNRIs in figure 2.
 Figure 2: Timeline of approved SNRIs.
Figure 2: Timeline of approved SNRIs.Mechanism of action
Monoamines are connected to the pathophysiology of depression. Symptoms are thought to appear because concentrations of neurotransmitters for example norepinephrine and serotonin are insufficient [7][11]. Medications to treat symptoms of depression effect the transmission of serotonin, norepinephrine and/or dopamine [7]. Older and more unselective antidepressants like TCAs and MAOIs, act by inhibiting the reuptake or metabolism, of norepinephrine and/or serotonin in the brain which concludes higher neurotransmitters concentrations [11]. Antidepressants that have dual mechanisms of action, inhibit both serotonin and norepinephrine reuptake, in some cases with weak effect on dopamine reuptake [7]. Antidepressants have effects on variable neuronal receptors like muscarinic-cholinergic, α1- and α2-adrenergic, and H1-histaminergic receptors, and sodium channels in the cardiac muscle, leading to decreased cardiac conduction and cardiotoxicity. Selectivity of antidepressant agents are based on the neurotransmitters that are thought to influence symptoms of depression [12]. Drugs that selectively block the reuptake of serotonin and/or norepinephrine have shown to be effective in treating depression and are better tolerated than TCAs. TCAs have comprehensive effects on various neurotransmitters receptors, which leads to lack of tolerability and increased risk of toxicity [13]
Tricyclic antidepressants
 Figure 3: Inhibiting the reuptake transport protein results in increased concentrations of serotonin and norepinephrine in the synaptic clefts, leading to improvement of depression symptoms.
Figure 3: Inhibiting the reuptake transport protein results in increased concentrations of serotonin and norepinephrine in the synaptic clefts, leading to improvement of depression symptoms.TCAs were the first medications that had dual mechanism of action. The mechanism of action of tricyclic secondary amine antidepressants is only partly understood. TCAs have dual inhibition effects on norepinephrine reuptake transporters and serotonin reuptake transporters. Increased norepinephrine and serotonin concentrations are obtained by inhibiting both of these transporter proteins. TCAs have substantially more affinity for norepinephrine reuptake proteins than the SSRIs. This is because of a formation of secondary amine TCA metabolites [14][15].
In addition, the TCAs interact with adrenergic receptors. This interaction seems to be critical for increased availability of norepinephrine in or near the synaptic clefts. Actions of imipramine-like tricyclic antidepressants have complex, secondary adaptions to their initial and sustained actions as inhibitors of norepinephrine transport and variable blockade of serotonin transport. Norepinephrine interacts with postsynaptic α- and β-adrenergic receptor subtypes and presynaptic α2-autoreceptors. The α2-receptors include presynaptic autoreceptors which limit the neurophysiological activity of noradrenergic neurons in the central nervous system. Formation of norepinephrine is reduced by autoreceptors through the ratelimiting enzyme tyrosine hydroxylase, by decreasing the expression of cyclic AMP-mediated phosphorylation-activation of the enzyme [15]. α2-receptors also cause decreased intracellular cyclic AMP expression which is resulted in smooth muscle relaxation or decreased secretion [16]. TCAs activate a negative-feedback mechanism through its effects on presynaptic receptor mediation. Probable explanation of effects on decreased neurotransmitter release is that as the receptors activate, inhibition of neurotransmitter release occurs, including suppression of voltage-gated Ca2+ currents and activation of G protein-coupled receptor-operated K+ currents. Repeated exposure of agents with this type of mechanism leads to inhibition of neurotransmitter release. However, repeated administration of TCAs finally results in decreased responses of α2-receptors. This desensitization of these respones may be caused due to increased exposure to the endogen norepinephrine or from prolonged occupation of the norepinephrine transport itself caused by allosteric effect. This adaptation allows the presynaptic synthesis and secration of norepinephrine to return to, or even exceed, normal levels of norepinephrine in the synaptic clefts. Overall inhibition of norepinephrine reuptake induced by TCAs, lead to decreased rates of neuron firing (mediated through α2-autoreceptors), metabolic activity, and release of neurotransmitters [15]. For schematic overview see figure 3.
TCAs do not block dopamine transport but might facilitate dopamine effects indirectly by transport-inhibition of dopamine into noreadrenergic terminals in cerebral cortex [15]. Because of multiple effects on different receptors, TCAs have poor tolerability which could result in adverse effects and increased risk of toxicity [13].
Selective serotonin reuptake inhibitors
Selective serotonin reuptake inhibitors (SSRIs) selectively inhibit the reuptake of serotonin and are a widely used group of antidepressants [17]. With increased receptors selectivity compared to TCAs, undesired effects like poor tolerability are avoided [15]. Serotonin is synthesized from an amino acid called L-tryptophan. Active transport system regulates the uptake of tryptophan across the blood-brain barrier. Serotonergic pathways are classified into two main ways in the brain; the ascending projections from the medial and dorsal raphe and the descending projections from the caudal raphe into the spinal cord. The reuptake blocking of SSRIs into the presynaptic terminal results in increased concentration of serotonin in the synaptic cleft, just like the TCAs, but with much less additional effects. Altering the serotonin concentrations to higher levels influence symptoms of anxiety which coexist with depression, leading to improvement of depression symptoms [17].
Selective norepinephrine reuptake inhibitors
There are two major regions in the brain where noradrenergic neurons are located. These regions are called locus coeruleus and the lateral tegmental. With administration of selective NRIs, neuronal activity in locus coeruleus region is induced because of increased concentration of norepinephrine in the synaptic cleft. This results in activation of α2-adrenoreceptors [11], as discussed in section 5.1. Assays have shown that selective NRIs have insignificant penchant for muscarinic-cholinergic, α1- and α2-adrenergic or H1-histaminergic receptors [12].
Dual serotonin and norepinephrine reuptake inhibitors
Agents with dual serotonin and norepinephrine reuptake inhibition (SNRIs) are sometimes called nontricyclic serotonin and norepinephrine reuptake inhibitors. Clinical studies suggest that compounds which increase the concentration in the synaptic cleft of both norepinephrine and serotonin are more successful than single acting agents in the treatment of depression. Dual reuptake inhibitors have low affinity at neuronal receptors of the other neurotransmitters which results in a low adverse effects, compared with the TCAs. Nontricyclic antidepressants have improved potency and onset action acceleration in antidepressant response than SSRI alone, which give the impression that synergism is an efficient property in mediating antidepressant activity. Between the nontricyclic SNRIs there are several important differences that are based on pharmacokinetics, metabolism to active metabolites, inhibition of CYP isoforms, effect of drug-drug interactions and the half-life of the nontricyclic SNRIs [14][18]. Combination of mechanisms of action in a single active agent is an important development in psychopharmacology [18].
Structure activity relationship (SAR)
Aryloxypropanamine scaffold
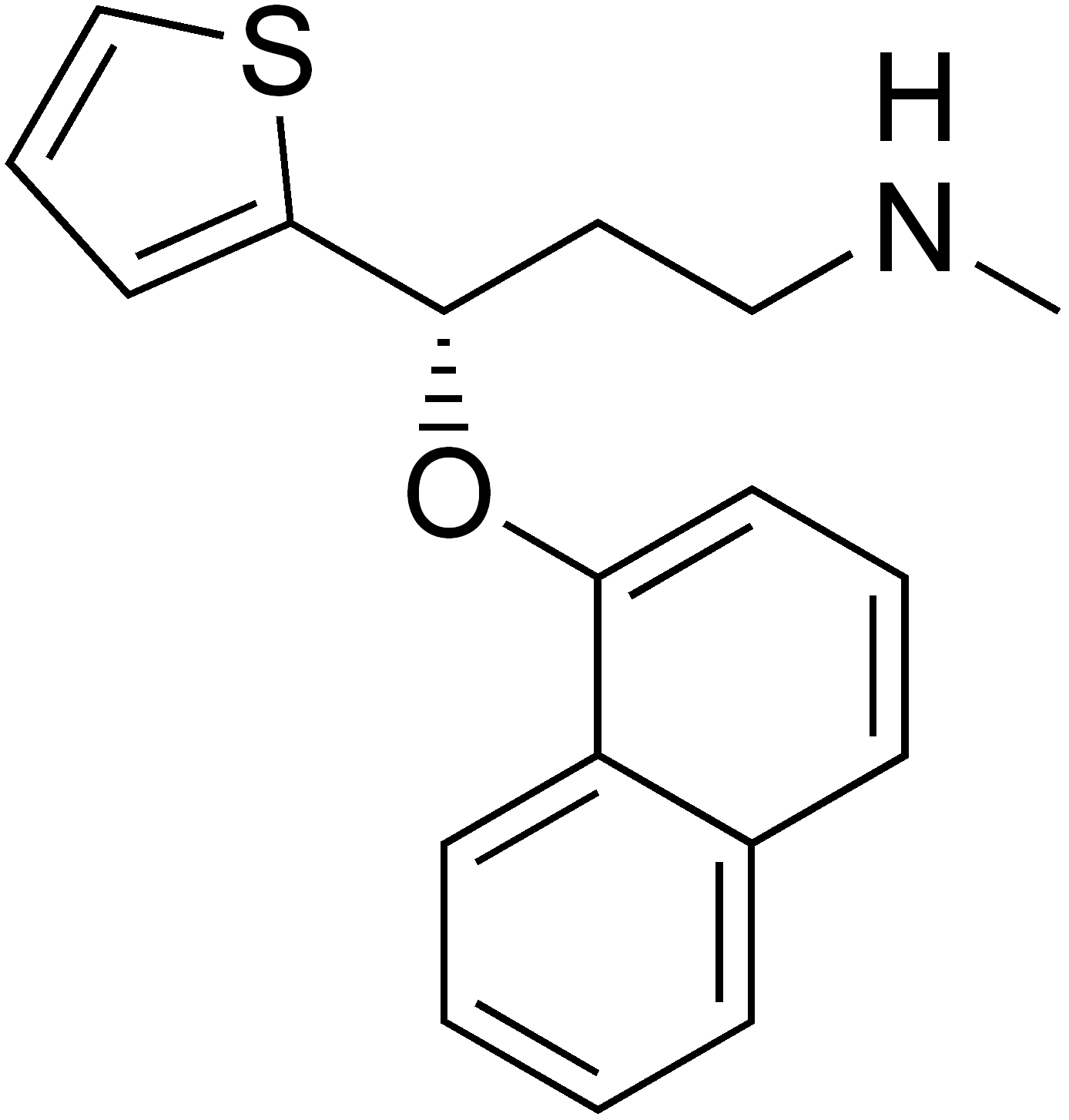
Atomoxetine, fluoxetine, duloxetine and reboxetine contain an aryloxypropanamine scaffold (see figure 4). This structural motif has potential for high affinity binding to biogenic amine transports [18]. Drugs containing an aryloxypropanamine scaffold have selectivity profile for norepinephrine and serotonin transporters that depends on the substitution pattern of the aryloxy ring. Selective NRIs contain a substituent in 2‘- position of the aryloxy ring but SSRIs contain a substituent in 4‘- position of the aryloxy ring. Atomoxetine, nisoxetine and reboxetine all have a substitution group in the 2´-position and are selective NRIs while compounds that have a substitution group in the 4´-position like fluoxetine and paroxetine are SSRIs. Drugs like duloxetine contain a phenyl group fused at the 2´ and 3´-positions. Duloxetine has therefore dual selective norepinephrine and serotonin reuptake inhibitory effects and has similar potencies for the both transporters [19]. The nature of the aromatic substituent R has a significant influence on the activity and selectivity of the compounds as inhibitors of the serotonin or the norepinephrine transporters [18]. See figure 5.
 Figure 4: Aryloxypropanamine scaffold evolution.
Figure 4: Aryloxypropanamine scaffold evolution.
Duloxetine 
Atomoxetine 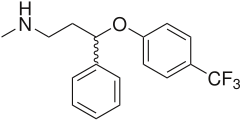
Fluoxetine 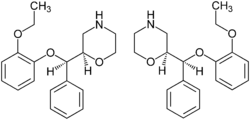
Reboxetine Figure 5: Agents with aryloxypropanamine scaffold.Cycloalkanol ethylamine scaffold
Venlafaxine contains a cycloalkanol ethylamine scaffold. Increasing the electron-withdrawing nature of the aromatic ring provides more potent inhibitory effect of norepinephrine uptake and improves the selectivity for norepinephrine over the serotonin transporter [20]. Effects of chloro, methoxy and trifluoromethyl substituents in the aromatic ring of cycloalkanol ethylamine scaffold were tested. The results showed that the strongest electron-withdrawing m-trifluoromethyl analogue exhibited the most potent inhibitory effect of norepinephrine and the most selectivity over serotonin uptake [20]. See agents with cycloalkanol ethylamine scaffolds in figure 6.

Cycloalkanol ethylamine scaffold 
R-substituent in cycloalkanol ethylamine scaffold 
Venlafaxine - Cycloalkanol ethylamine scaffold (in red) Figure 6: Agents with cycloalkanol ethylamine scaffoldMilnacipran
Milnacipran is structurally different from other SNRIs [14]. The SAR of milnacipran derivatives at transporter level is still largely unclear and is based on in vivo efficacy that was reported in 1987. N-methylation of milnacipran in substituent group R4 and R5 reduces the norepinephrine and serotonin activity [21]. Researches on different secondary amides in substitution groups R6 and R7 showed that π electrons play an important role in the interaction between transporters and ligands. A phenyl group in substituent R6 showed effect on norepinephrine transporters. Substituent groups in R6 and R7 with allylic double bond showed significant improved effect on both norepinephrine and serotonin transporters [21]. Studies have shown that when introducing a 2-methyl group in substituent R3 the potency at norepinephrine and serotonin transporters are almost abolished. Methyl group in substituent group R1 and R2 also abolished the potency at norepinephrine and serotonin transporters. Researchers found out that when replacing one of the ethyl groups of milnacipran with an allyl moiety increases the norepinephrine potency [22]. The pharmacophore of milnacipran derivatives is still largely unclear [21].
 Figure 7: Structure of milnacipran.
Figure 7: Structure of milnacipran.The conformation of milnacipran is an important part of its pharmacophore. Changing the SAR in milnacipran changes the stereochemistry of the compound and affects the norepinephrine and serotonin concentration. Milnacipran is marketed as a racemic mixture. Effects of milnacipran resides in the 1S,2R-isomer and substitution of the phenyl group in the 1S,2R isomer has negative impact on norepinephrine concentration [22]. Milnacipran has low molecular weight and low lipophilicity. Because of these properties milnacipran exhibits almost ideal pharmacokinetics in humans such as high bioavailability, low inter-subject variability, limited liver enzyme interaction, moderate tissue distribution and a reasonably long elimination half-life. Milnacipran´s lack of drug-drug interactions via cytochrome P450 enzymes is thought to be an attractive feature because many of the central nervous system drugs are highly lipophilic and are mainly eliminated by liver enzymes [22].
Future development of SAR
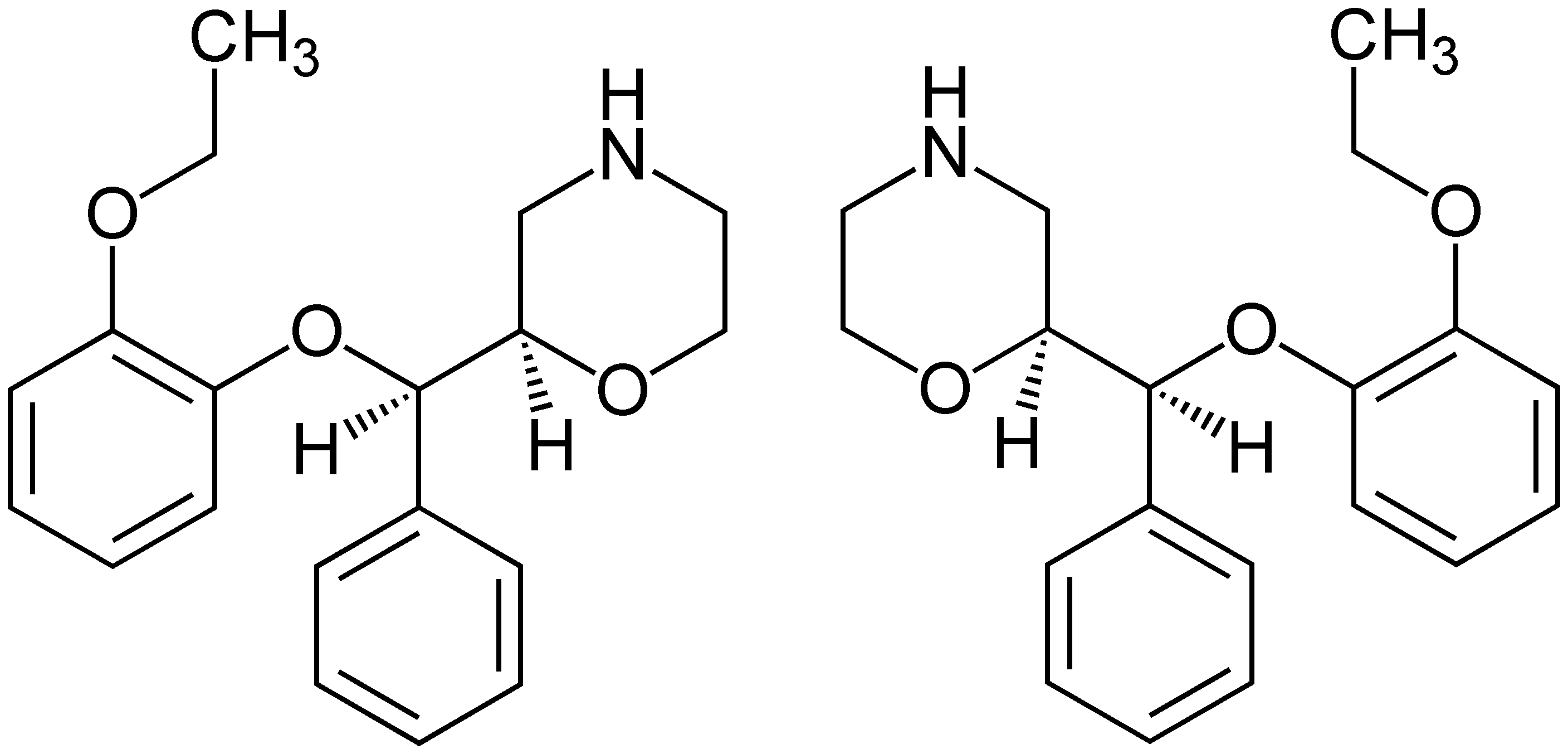
The application of an aryloxypropanamine scaffold has generated a number of potent MAOIs [23]. Before the development of duloxetine, the exploration of aryloxypropanamine SAR resulted in the identification of fluoxetine and atomoxetine. The same motif can be found in reboxetine where it is constrained in a morpholine ring system. Some studies have been made where the oxygen in reboxetine is replaced by sulfur to give arylthiomethyl morpholine. Some of the arylthiomethyl morpholine derivatives maintain potent levels of serotonin and norepinephrine reuptake inhibition. Dual serotonin and norepinephrine reuptake inhibition resides in different enantiomers for arylthiomethyl morpholine scaffold [24]. Possible drug candidates with dual serotonin and norepinephrine reuptake inhibitory activity have also been derived from piperazine, 3-amino-pyrrolidine and benzylamine templates [25].
Clinical trials
Several studies have shown that antidepressant drugs which have combined serotonergic and noradrenergic activity is more effective than SSRIs in treating MDD. Results showed that serotonergic-noradrenergic antidepressant drugs seem to have a modest efficacy advantage compared to SSRIs in treating MDD [26] and have less side effects[27]. Serotonergic-noradrenergic antidepressant drugs used to treat MDD compared to SSRIs and mirtazapine seem to be less expensive than other drugs for example in the UK. Further research is needed to examine whether larger differences between classes of antidepressant drugs might exist in specific MDD sub-populations or for specific MDD symptoms[28].
Data from clinical trials have indicated that SNRIs might have pain relieving properties. The pain perception and transmission in the central nervous system have not been fully elucidated however, extensive data support a role of serotonin and norepinephrine in the modulation of pain. This is also supported by data from clinical trials in humans in which antidepressants have been shown to reduce pain and functional impairment in central and neuropathic pain conditions. This property of SNRIs might be used to reduce doses of other pain relieving medication and lower frequency of limited efficacy, safety and tolerability issues[29]. Clinical research data have shown in patients with GAD that the SNRI duloxetine is significantly more effective than placebo in reducing painful physical symptoms of GAD after short-term and long-term treatment. Findings suggested that painful physical symptoms reoccur with relapsing that indicate a need for ongoing treatment in patients with GAD and concurrent painful physical symptoms[30].
Current status
Levomilnacipran is the enantiomer of milnacipran. Like milnacipran, levomilnacipran is a selective SNRI. Levomilnacipran is currently under development for the treatment of MDD[31].
Bicifadine is a SNRI. Bicifadine is being developed for the treatment of acute and chronic pain. Clinical research have shown that bicifadine is effective in the treatment of acute dental pain and bunionectomy pain. Bicifadine has also been reported to be as effective as the standard of care in reducing chronic lower back pain[32].
Products of SNRIs
Active ingredient Product name Therapeutic use Binding affinity
(Ki nm)Structure Venlafaxine Efexor® Effexor® Treatment of treatment-resistant depression (TRD), generalized anxiety disorder (GAD), chronic pain syndromes [4] 5-HT:7,8
NE:1920
D:6050 [13]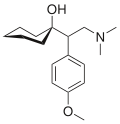
Milnacipran Savella® Ixel® Dalcipran® Toledomin® Treatment of pain and multiple symptoms associated with fibromyalgia syndrome, treatment of MDD [10] 5-HT:8,4
NE:22
D:>100000 [13]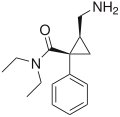
Duloxetine Cymbalta® Ariclaim® Xeristar® Yentreve® Treatment of MDD and fibromyalgia [7] 5-HT:0,07
NE:1,2
D:230 [13]
Levomilnacipran Not listed Under development [31] 5-HT:NA
NE:NA
D:NA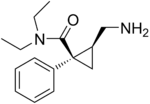
Bicifadine Not listed Under development [32] 5-HT:NA
NE:NA
D:NA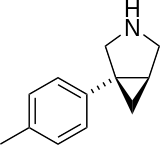
Desvenlafaxine Pristiq® Treatment of MDD in adults [8] 5-HT:NA
NE:NA
D:NA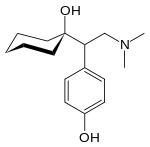
Sibutramine Meridia® Reductil® Treatment of obesity [6] 5-HT:NA
NE:NA
D:NA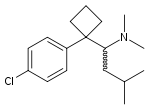
5-HT: Serotonin - NE: Norepinephrine - D: Dopamine - NA: Not available
Abbreviations
- Serotonin–norepinephrine reuptake inhibitors (SNRI)
- Major depressive disorder (MDD)
- Serotonin (5-Hydroxytryptamine, 5-HT)
- Norepinephrine (NE, noradrenalin)
- Serotonin transporter (SERT)
- Norepinephrine transporter (NET)
- Selective serotonin reuptake inhibitors (SSRI)
- Norepinephrine reuptake inhibitors (NRI)
- Tricyclic antidepressants (TCA)
- Monoamine oxidase inhibitors (MAOI)
- Food and Drug Administration (FDA)
- Treatment-resistant depression (TRD)
- Generalized anxiety disorder (GAD)
- Structure activity relationship (SAR)
See also
- Serotonin (5-HT)
- Norepinephrine (NE)
- Reuptake inhibitor (RI)
- Norepinephrine reuptake inhibitor (NRI)
- Serotonin reuptake inhibitor (SRI)
- Selective serotonin reuptake inhibitor (SSRI)
- Serotonin–norepinephrine reuptake inhibitor (SNRI)
- Dopamine reuptake inhibitor (DRI)
- Serotonin–norepinephrine–dopamine reuptake inhibitor (SNDRI)
- Norepinephrine-dopamine reuptake inhibitor (NDRI)
References
- ^ Cashman, J. R. and Ghirmai, S. (2009). Inhibition of serotonin and norepinephrine reuptake and inhibition of phosphodiesterase by multi-target inhibitors as potential agents for depression. Bioorganic & Medicinal Chemistry, 17, 6890–6897.PubMed
- ^ Spina, E., Santoro, V. and D'Arrigo, C. (2008). Clinically Relevant Pharmacokinetic Drug Interactions with Second-Generation Antidepressants: An Update.Clinical Therapeutics, 30(7),1206-1227.PubMed
- ^ a b Lieberman, J. A. (2003). History of the Use of Antidepressants in Primary Care. Primary Care Companion J Clin Psychiatry, 5(7), 6-10.
- ^ a b c Gutierrez, M. A., Stimmel, G. L. and Aiso, J. Y. (2003). Venlafaxine: A 2003 Update. Clinical Therapeutics, 25(8), 2138-2154.PubMed
- ^ a b Ruelas, E. G., Diaz-Martinez, A., Ruiz, R. M. and the venlapaxine-global awareness program study collaborative group. (1997). An open assessment of the acceptability, efficacy and tolerance of venlafaxine in usual care settings. Current therapeutic research, 58(9), 609-630.doi:10.1016/S0011-393X(97)80088-4
- ^ a b Luque, C. A., & Rey, J. A. (2002). The discovery and status of sibutramine as an anti-obesity drug. European Journal of Pharmacology, 440(2-3), 119-128.PubMed
- ^ a b c d e Hunziker, M.E. et al. (2005). Duloxetine Hydrochloride: A New Dual-Acting Medication for the Treatment of Major Depressive Disorder. Clinical Therapeutics, 27, 8.PubMed
- ^ a b Perry, R. and Cassagnol, M. (2009). Desvenlafaxine: A New Serotonin-Norepinephrine Reuptake Inhibitor for the Treatment of Adults With Major Depressive Disorder. Clinical Therapeutics, 31, 1374-1404.PubMed
- ^ Clauw, D. J., Mease, P., Palmer, R. H., Gendreau, M., & Wang, Y. (2008). Milnacipran for the Treatment of Fibromyalgia in Adults: A 1S-Week, Multicenter, Randomized, Double-Blind, Placebo-Controlled, Multiple-Dose Clinical Trial. Clinical Therapeutics, 30 (11), 1988-2004.PubMed
- ^ a b Morishita, S., & Arita, S. (2003). The clinical use of milnacipran for depression. European Psychiatry, 18 (1), 34-35.doi:10.1016/S0924-9338(02)00003-2
- ^ a b c Grandoso, L., Pineda, J. and Ugedo, L. (2003). Comparative study of the effects of desipramine and reboxetine on locus coeruleus neurons in rat brain slices. Neuropharmacology, 46, 815-823.PubMed
- ^ a b Brunello, N. et al. (2002). The role of noradrenaline and selective noradrenaline reuptake inhibition in depression. European Neuropsychopharmacology, 12, 461-475.PubMed
- ^ a b c d e Stahl, S.M., Grady, M.M., Moret, C. and Briley, M. (2005). SNRIs: Their Pharmacology, Clinical Efficacy, and Tolerability in Comparison with Other Classes of Antidepressants. CNS Spectrums. 10, 9, 732-747.PubMed
- ^ a b c Lemke, T. L., Williams, D.A., Roche, V.F. and Zito, S. W. (2008). Foye´s principles of medicinal chemistry (6. edition) (p. 547-67; 581-582). USA: Lippincott Williams & Wilkins
- ^ a b c d e Brunton, L.L., Lazo, J.S. and Parker, K.L. (Editors). (2006). Goodman & Gilman’s: The Pharmacological Basis of Therapeutics. 11. edition. New York: McGraw-Hill
- ^ Silverthorn, D.U. (Editor). (2007). Human Physiology. 4. edition. (pages 383-384) San Francisco: Pearson
- ^ a b Nutt, D.J. et al. (1999). Mechanisms of action of SSRIs in the treatment of psychiatric disorders. European Neurophsychopharmacology,9, 3, 81-86.doi:10.1016/S0924-977X(99)00030-9
- ^ a b c d Boot, J., Cases, M., Clark, B. P., Findlay, J., Gallagher, P. T., Hayhurst, L., et al. (2005). Discovery and structure-activity relationships of novel selective norepinephrine and dual SNRIs. Bioorganic & Medicinal Chemistry Letters, 15(3), 699-703.PubMed
- ^ Mahaney, P. E., Vu, A. T., McComas, C. C., Zhang, P., Nogle, L. M., Watts, W. L., Sarkahian, A., Leventhal, L., Sullivan, N. R., Uveges A. J. and Trybulski, E. J. (2006). Synthesis and activity of a new class of dual acting norepinephrine and serotonin reuptake inhibitors: 3-(1H-indol-1-yl)-3-arylpropan-1-amines. Bioorganic & Medicinal Chemistry, 14 (24), 8455-8466.PubMed
- ^ a b Mahaney, P. E., Gavrin, L. K., Trybulski, E. J., Stack, G. P., Vu, A. T., Cohn, S. T., et al. (2008). Structure-activity relationships of the cycloalkanol ethylamine scaffold: Discovery of selective norepinephrine reuptake inhibitors. Journal of Medicinal Chemistry, 51(13), 4038-4049.PubMed
- ^ a b c Chen, C., Dyck, B., Fleck, B. A., Foster, A. C., Grey, J., Jovic, F., et al. (2008). Studies on the SAR and pharmacophore of milnacipran derivatives as monoamine transporter inhibitors. Bioorganic & Medicinal Chemistry Letters, 18(4).doi:10.1016/j.bmcl.2008.01.011
- ^ a b c Tamiya, J., Dyck, B., Zhang, M. Z., Phan, K., Fleck, B. A., Aparicio, A., et al. (2008). Identification of 1S, 2R-milnacipran analogs as potent norepinephrine and serotonin transporter inhibitors. Bioorganic & Medicinal Chemistry Letters, 18(11), 3328-3332.doi:10.1016/j.bmcl.2008.04.025
- ^ Vu, A. T., Cohn, S. T., Terefenko, E. A., Moore, W. J., Zhang, P., Mahaney, P. E., et al. (2009). 3-(Arylamino)-3-phenylpropan-2-olamines as a new series of dual norepinephrine and serotonin reuptake inhibitors. Bioorganic & Medicinal Chemistry Letters, 19(9), 2464-2467.doi:10.1016/j.bmcl.2009.03.054 PMID 19329313
- ^ Boot, J. R., Brace, G., Delatour, C. L., Dezutter, N., Fairhurst, J., Findlay, J., et al. (2004). Benzothienyloxy phenylpropanamines, novel dual inhibitors of serotonin and norepinephrine reuptake. Bioorganic & Medicinal Chemistry Letters, 14(21), 5395-5399.PubMed
- ^ Fish, P. V., Deur, C., Gan, X., Greene, K., Hoople, D., Mackenny, M., et al. (2008). Design and synthesis of morpholine derivatives. SAR for dual serotonin & noradrenaline reuptake inhibition. Bioorganic & Medicinal Chemistry Letters, 18(8), 2562-2566.doi:10.1016/j.bmcl.2008.03.050
- ^ Papakostas, G. I., Thase, M. E., Maurizio, F., Nelson, C. J., & Shelton, R. C. (2007). Are Antidepressant Drugs That Combine Serotonergic and Noradrenergic Mechanisms of Action More Effective Than the Selective Serotonin Reuptake Inhibitors in Treating Major Depressive Disorder? A Meta-analysis of Studies of Newer Agents. Biological Psychiatry, 62 (11), 1217-1227.PubMed
- ^ Nemeroff, C. B., Thase, M. E., & Group, E. 0. (2007). A double-blind, placebo-controlled comparison of venlafaxine and fluoxetine treatment in depressed outpatients. Original Research Article Journal of Psychiatric Research, 41 (3-4), 351-359.doi:10.1016/j.jpsychires.2005.07.009
- ^ Benedict, Á., Arellano, J., De Coc, E., & Baird, J. (2010). Economic evaluation of duloxetine versus serotonin selective reuptake inhibitors and venlafaxine XR in treating major depressive disorder in Scotland. Scotland. Journal of affective disorders, 120 (1-3), 94-104.PubMed
- ^ Marks, D. M., Shah, M. J., Patkar, A. A., Masand, P. S., Park, G.-Y., & Pae, C.-U. (2009). Serotonin-Norepinephrine Reuptake Inhibitors for Pain Control. Current Neuropharmacology, 7 (4), 331-336.doi:10.2174/157015909790031201
- ^ Beesdo, K., Hartford, J., Russell, J., Spann, M., Ball, S., & Wittchen, H.-U. (2009). The short- and long-term effect of duloxetine on painful physical symptoms in patients with generalized anxiety disorder: Results from three clinical trials. Journal Anxiety Disorders, 23 (8), 1064-1071.PubMed
- ^ a b Lecrubier, Y., Mansuy, L., Bose, A., Li, J., & Werner, P. (2010). Efficacy of levomilnacipran in improving symptoms and functional impairment associated with major depressive disorder. European Neuropsychopharmacology, 20 (3), 390.doi:10.1016/S0924-977X(10)70553-8
- ^ a b Zhang, M., Jovic, F., Vickers, T., Dyck, B., Tamiya, J., Gray, J., et al. (2008). Studies on the structure–activity relationship of bicifadine analogs as monoamine transporter inhibitors. Bioorganic & Medicinal Chemistry Letters, 18 (13), 3682-3686.doi:10.1016/j.bmcl.2008.05.077
Drug design steps in design Case studies of discovery and development of drug classes triptans · cephalosporins · antiandrogens · Bcr-Abl tyrosine kinase inhibitors · nucleoside and nucleotide reverse transcriptase inhibitors · melatonin receptor agonists · proton pump inhibitors · angiotensin receptor blockers · dual serotonin and norepinephrine reuptake inhibitors · cyclooxygenase 2 inhibitors · TRPV1 antagonistsCategories:- Drug discovery
- Antidepressants









Wikimedia Foundation. 2010.