- Androgen insensitivity syndrome
-
Androgen insensitivity syndrome Classification and external resources 
AIS results when the function of the androgen receptor (AR) is impaired. The AR protein (pictured) mediates the effects of androgens in the human body.ICD-10 E34.5 ICD-9 259.5 OMIM 312300 300068 DiseasesDB 29662 12975 eMedicine ped/2222 MeSH D013734 GeneReviews Androgen Insensitivity Syndrome  Women with AIS and related DSD conditions
Women with AIS and related DSD conditions
Androgen insensitivity syndrome (AIS) is a condition that results in the partial or complete inability of the cell to respond to androgens.[1][2][3] The unresponsiveness of the cell to the presence of androgenic hormones can impair or prevent the masculinization of male genitalia in the developing fetus, as well as the development of male secondary sexual characteristics at puberty, but does not significantly impair female genital or sexual development.[3][4] As such, the insensitivity to androgens is only clinically significant when it occurs in genetic males (i.e. individuals with a Y-chromosome, or more specifically, an SRY gene).[1] Clinical phenotypes in these individuals ranges from a normal male habitus with mild spermatogenic defect or reduced secondary terminal hair, to a full female habitus, despite the presence of a Y-chromosome.[1][5][6][7][8][9]
AIS is divided into three categories that are differentiated by the degree of genital masculinization: complete androgen insensitivity syndrome (CAIS) is indicated when the external genitalia is that of a normal female, mild androgen insensitivity syndrome (MAIS) is indicated when the external genitalia is that of a normal male, and partial androgen insensitivity syndrome (PAIS) is indicated when the external genitalia is partially, but not fully masculinized.[1][2][5][6][7][10][11][12][13]
Androgen insensitivity syndrome is the largest single entity that leads to 46,XY undermasculinized genitalia.[14]
Contents
Signs and symptoms
AIS is broken down into three classes based on phenotype: complete androgen insensitivity syndrome (CAIS), partial androgen insensitivity syndrome (PAIS), and mild androgen insensitivity syndrome (MAIS).[1][2][5][6][7][10][11][12][13] A supplemental system of phenotypic grading that uses seven classes instead of the traditional three was proposed by pediatric endocrinologist Charmian A. Quigley et al. in 1995.[3] The first six grades of the scale, grades 1 through 6, are differentiated by the degree of genital masculinization; grade 1 is indicated when the external genitalia is fully masculinized, grade 6 is indicated when the external genitalia is fully feminized, and grades 2 through 5 quantify four degrees of decreasingly masculinized genitalia that lie in the interim.[3] Grade 7 is indistinguishable from grade 6 until puberty, and is thereafter differentiated by the presence of secondary terminal hair; grade 6 is indicated when secondary terminal hair is present, whereas grade 7 is indicated when it is absent.[3] The Quigley scale can be used in conjunction with the traditional three classes of AIS to provide additional information regarding the degree of genital masculinization, and is particularly useful when the diagnosis is PAIS.[2][15]
Complete AIS
Main article: Complete androgen insensitivity syndromePartial AIS
Main article: Partial androgen insensitivity syndromeMild AIS
Main article: Mild androgen insensitivity syndromeGenetics
 Location and structure of the human androgen receptor. Top, The AR gene is located on the proximal long arm of the X chromosome. Middle, The eight exons are separated by introns of various lengths. Bottom, Illustration of the AR protein, with primary functional domains labeled (not representative of actual 3-D structure).[3]
Location and structure of the human androgen receptor. Top, The AR gene is located on the proximal long arm of the X chromosome. Middle, The eight exons are separated by introns of various lengths. Bottom, Illustration of the AR protein, with primary functional domains labeled (not representative of actual 3-D structure).[3]The human androgen receptor (AR) is a protein encoded by a gene located on the proximal long arm of the X chromosome (locus Xq11-Xq12).[16] The protein coding region consists of approximately 2,757 nucleotides (919 codons) spanning eight exons, designated 1-8 or A-H.[1][3] Introns vary in size between 0.7 and 26 kb.[3] Like other nuclear receptors, the androgen receptor protein consists of several functional domains: the transactivation domain (also called the transcription-regulation domain or the amino / NH2-terminal domain), the DNA-binding domain, the hinge region, and the steroid-binding domain (also called the carboxyl-terminal ligand-binding domain).[1][2][3][13] The transactivation domain is encoded by exon 1, and makes up more than half of the AR protein.[3] Exons 2 and 3 encode the DNA-binding domain, while the 5' portion of exon 4 encodes the hinge region.[3] The remainder of exon 4 through exon 8 encodes the ligand binding domain.[3]
Trinucleotide satellite lengths and AR transcriptional activity
The androgen receptor gene contains two polymorphic trinucleotide microsatellites in exon 1.[2] The first microsatellite (nearest the 5' end) contains 8 [17] to 60 [18][19] repetitions of the glutamine codon "CAG" and is thus known as the polyglutamine tract.[3] The second microsatellite contains 4 [20] to 31 [21] repetitions of the glycine codon "GGC" and is known as the polyglycine tract.[22] The average number of repetitions varies by ethnicity, with Caucasians exhibiting an average of 21 CAG repeats, and Blacks 18.[23] In men, disease states are associated with extremes in polyglutamine tract length; prostate cancer,[24] hepatocellular carcinoma,[25] and mental retardation [17] are associated with too few repetitions, while spinal and bulbar muscular atrophy (SBMA) is associated with a CAG repetition length of 40 or more.[26] Some studies indicate that the length of the polyglutamine tract is inversely correlated with transcriptional activity in the AR protein, and that longer polyglutamine tracts may be associated with male infertility [27][28][29] and undermasculinized genitalia in men.[30] However, other studies have indicated that no such correlation exists.[31][32][33][34][35][36] A comprehensive meta-analysis of the subject published in 2007 supports the existence of the correlation, and concluded that these discrepancies could be resolved when sample size and study design are taken into account.[11] Some studies suggest that longer polyglycine tract lengths are also associated with genital masculinization defects in men.[37][38] Other studies find no such association.[39]
AR mutations
As of 2010, over 400 AR mutations have been reported in the AR mutation database, and the number continues to grow.[2] Inheritance is typically maternal and follows an X-linked recessive pattern;[1][40] individuals with a 46,XY karyotype will always express the mutant gene since they only have one X chromosome, whereas 46,XX carriers will be minimally affected. 30% of the time, the AR mutation is a spontaneous result, and is not inherited.[10] Such de novo mutations are the result of a germ cell mutation or germ cell mosaicism in the gonads of one of the parents, or a mutation in the fertilized egg itself.[41] In one study,[42] it was found that 3 out of 8 de novo mutations occurred in the post-zygotic stage, leading to the estimate that up to one third of de novo mutations result in somatic mosaicism.[1] It is worthwhile to note that not every mutation of the AR gene results in androgen insensitivity; one particular mutation occurs in 8 to 14 percent of genetic males,[43][44][45][46] and is thought to adversely affect only a small number of individuals when other genetic factors are present.[47]
Other causes
Some individuals with CAIS or PAIS do not have any AR mutations despite clinical, hormonal, and histological features sufficient to warrant an AIS diagnosis; up to 5% of women with CAIS do not have an AR mutation,[2] as well as between 27% [6][48] and 72% [49] of individuals with PAIS.
In one patient, it was shown that the underlying cause for presumptive PAIS was a mutant steroidogenic factor-1 (SF-1) protein.[50] In another patient, it was shown that CAIS was the result of a deficit in the transmission of a transactivating signal from the N-terminal region of the normal androgen receptor to the basal transcription machinery of the cell.[51] It was suggested that a coactivator protein interacting with the activation function 1 (AF-1) transactivation domain of the androgen receptor was deficient in this patient.[51] The signal disruption could not be corrected by supplementation with any coactivators known at the time, nor was the absent coactivator protein characterized, which left some in the field unconvinced that a mutant coactivator would explain the mechanism of androgen resistance in CAIS or PAIS patients with a normal AR gene.[1]
XY karyotype
Depending on the mutation, a person with a (46,XY karyotype) and AIS can have either a male (MAIS) or female (CAIS) phenotype,[52] or may have genitalia that is only partially masculinized (PAIS).[53] The gonads are testes regardless of phenotype due to the influence of the Y-chromosome.[54][55] A 46,XY female thus does not have ovaries or a uterus,[56] and can neither contribute an egg towards conception nor gestate a child.
Several case studies of fertile 46,XY males with androgen insensitivity have been published,[4][57][58][59][60] although this group is thought to be a minority.[13] Additionally, some infertile males with MAIS have been able to conceive children after increasing their sperm count through the use of supplementary testosterone.[1][61] A genetic male conceived by a man with androgen insensitivity would not receive his father's X chromosome, and thus would neither inherit nor carry the gene for the syndrome. A genetic female conceived in such a way would receive her father's X chromosome, and would thus become a carrier.
XX karyotype
Genetic females (46,XX karyotype) have two X chromosomes, and thus have two AR genes. A mutation in one (but not both) of the AR genes results in a minimally affected, fertile, female carrier. Some carriers have been noted to have slightly reduced body hair, delayed puberty, and / or tall stature, presumably due to skewed X-inactivation.[3][4] A female carrier will pass the affected AR gene to her children 50% of the time. If the affected child is a genetic female, she too will be a carrier. An affected 46,XY child will have androgen insensitivity syndrome.
A genetic female with mutations in both AR genes could theoretically result from the union of a fertile man with androgen insensitivity and a female carrier of the gene, or from de novo mutation. However, given the scarcity of fertile androgen insensitive men and low incidence of AR mutation, the chances of this occurrence is small. The phenotype of such an individual is a matter of speculation; as of 2010, no such documented case has been published.
Correlation of genotype and phenotype
Individuals with partial androgen insensitivity, unlike those with the complete or mild forms, present at birth with ambiguous genitalia, and the decision to raise the child as male or female is often not obvious.[1][41][62] Unfortunately, it is often the case that little information regarding phenotype can be gleaned from precise knowledge of the AR mutation itself; it is well established that the same AR mutation may cause significant variation in the degree of masculinization in different individuals, even among members of the same family.[1][40][53][63][64][65][66][67][68][69] Exactly what causes this variation is not entirely understood, although factors contributing to it could include the lengths of the polyglutamine and polyglycine tracts,[70] sensitivity to and variations in the intrauterine endocrine milieu,[53] the effect of coregulatory proteins that are active in Sertoli cells,[22][71] somatic mosaicism,[1] expression of the 5RD2 gene in genital skin fibroblasts,[63] reduced AR transcription and translation from factors other than mutations in the AR coding region,[72] an unidentified coactivator protein,[51] enzyme deficiencies such as 21-hydroxylase deficiency,[4] or other genetic variations such as a mutant steroidogenic factor-1 (SF-1) protein.[50] The degree of variation, however, does not appear to be constant across all AR mutations, and is much more extreme in some.[1][4][47][53] Missense mutations that result in a single amino acid substitution are known to produce the most phenotypic diversity.[2]
Pathophysiology
 Normal function of the androgen receptor. Testosterone (T) enters the cell and, if 5-alpha-reductase is present, is converted into dihydrotestone (DHT). Upon steroid binding, the androgen receptor (AR) undergoes a conformational change and releases heat shock proteins (hsps). Phosphorylation (P) occurs before or after steroid binding. The AR translocates to the nucleus where dimerization, DNA binding, and the recruitment of coactivators occur. Target genes are transcribed (mRNA) and translated into proteins.[3][13][19][73]
Normal function of the androgen receptor. Testosterone (T) enters the cell and, if 5-alpha-reductase is present, is converted into dihydrotestone (DHT). Upon steroid binding, the androgen receptor (AR) undergoes a conformational change and releases heat shock proteins (hsps). Phosphorylation (P) occurs before or after steroid binding. The AR translocates to the nucleus where dimerization, DNA binding, and the recruitment of coactivators occur. Target genes are transcribed (mRNA) and translated into proteins.[3][13][19][73]Androgens and the androgen receptor
Main article: Androgen receptorThe effects that androgens have on the human body --- virilization, masculinization, anabolism, etc. --- are not brought about by androgens themselves, but rather are the result of androgens bound to androgen receptors; the androgen receptor mediates the effects of androgens in the human body.[74] Likewise, under normal circumstances, the androgen receptor itself is inactive in the cell until androgen binding occurs.[3]
The following series of steps illustrates how androgens and the androgen receptor work together to produce androgenic effects:[1][2][3][13][19][75][76]
- Androgen enters the cell.
- Only certain organs in the body, such as the gonads and the adrenal glands, produce the androgen testosterone.
- Testosterone is converted into dihydrotestosterone, a chemically similar androgen, in cells containing the 5 alpha reductase enzyme.
- Both androgens exert their influence through binding with the androgen receptor.
- Androgen binds with the androgen receptor.
- The androgen receptor is expressed ubiquitously throughout the tissues of the human body.
- Before it binds with an androgen, the androgen receptor is bound to heat shock proteins.
- These heat shock proteins are released upon androgen binding.
- Androgen binding induces a stabilizing, conformational change in the androgen receptor.
- The two zinc fingers of the DNA-binding domain are exposed as a result of this new conformation.
- AR stability is thought to be aided by type II coregulators, which modulate protein folding and androgen binding, or facilitate NH2/carboxyl-terminal interaction.
- The hormone-activated androgen receptor is phosphorylated.
- Receptor phosphorylation can occur before androgen binding, although the presence of androgen promotes hyperphosphorylation.
- The biological ramifications of receptor phosphorylation are unknown.
- The hormone-activated androgen receptor translocates to the nucleus.
- Nucleocytoplasmic transport is in part facilitated by an amino acid sequence on the AR called the nuclear localization signal.
- The AR's nuclear localization signal is primarily encoded in the hinge region of the AR gene.
- Homodimerization occurs.
- Dimerization is mediated by the second (nearest the 3' end) zinc finger.
- DNA binding to regulatory androgen response elements occurs.
- Target genes contain (or are flanked by) transcriptional enhancer nucleotide sequences that interact with the first zinc finger.
- These areas are called androgen response elements.
- Coactivators are recruited by the AR.
- Type I coactivators (i.e., coregulators) are thought to influence AR transcriptional activity by facilitating DNA occupancy, chromatin remodeling, or the recruitment of general transcription factors associated with RNA polymerase II holocomplex.
- Target gene transcription ensues.
In this way, androgens bound to androgen receptors regulate the expression of target genes, and thus produce androgenic effects.
It is theoretically possible for certain mutant androgen receptors to function without androgens; in vitro studies have demonstrated that a mutant androgen receptor protein can induce transcription in the absence of androgen if its steroid binding domain is deleted.[77][78] Conversely, the steroid-binding domain may act to repress the AR transactivation domain, perhaps due to the AR's unliganded conformation.[3]
 Sexual differentiation. The human embryo has indifferent sex accessory ducts until the seventh week of development.[79]
Sexual differentiation. The human embryo has indifferent sex accessory ducts until the seventh week of development.[79]Androgens in fetal development
Human embryos develop similarly for the first six weeks, regardless of genetic sex (46,XX or 46,XY karyotype); the only way to tell the difference between 46,XX or 46,XY embryos during this time period is to look for Barr bodies or a Y-chromosome.[80] The gonads begin as bulges of tissue called the genital ridges at the back of the abdominal cavity, near the midline. By the fifth week, the genital ridges differentiate into an outer cortex and an inner medulla, and are called indifferent gonads.[80] By the sixth week, the indifferent gonads begin to differentiate according to genetic sex. If the karyotype is 46,XY, testes develop due to the influence of the Y chromosome’s SRY gene.[54][55] This process does not require the presence of androgen, nor a functional androgen receptor.[54][55]
Until approximately the seventh week of development, the embryo has indifferent sex accessory ducts, which consist of two pairs of ducts: the Müllerian ducts and the Wolffian ducts.[80] The testes secrete anti-Müllerian hormone around this time to suppress the development of the Müllerian ducts, and cause their degeneration.[80] Without this anti-Müllerian hormone, the Müllerian ducts develop into the female internal genitalia (uterus, cervix, fallopian tubes, and upper vaginal barrel).[80] Unlike the Müllerian ducts, the Wolffian ducts will not continue to develop by default.[81] In the presence of testosterone and functional androgen receptors, the Wolffian ducts develop into the epididymides, vasa deferentia, and seminal vesicles.[80] If the testes fail to secrete testosterone, or the androgen receptors do not function properly, the Wolffian ducts degenerate.[82]
 Masculinization of the male genitalia is dependent on both testosterone and dihydrotestosterone.[79]
Masculinization of the male genitalia is dependent on both testosterone and dihydrotestosterone.[79]Masculinization of the external genitalia (the penis, penile urethra, and scrotum), as well as the prostate, are dependent on the androgen dihydrotestosterone.[83][84][85][86] Testosterone is converted into dihydrotestosterone by the 5-alpha reductase enzyme.[87] If this enzyme is absent or deficient, then dihydrotestosterone will not be created, and the external male genitalia will not develop properly.[83][84][85][86][87] As is the case with the internal male genitalia, a functional androgen receptor is needed in order for dihydrotestosterone to regulate the transcription of target genes involved in development.[74]
Pathogenesis of Androgen Insensitivity Syndrome
Mutations in the androgen receptor gene can cause problems with any of the steps involved in androgenization, from the synthesis of the androgen receptor protein itself, through the transcriptional ability of the dimerized, androgen-AR complex.[3] AIS can result if even one of these steps is significantly disrupted, as each step is required in order for androgens to successfully activate the AR and regulate gene expression.[3] Exactly which steps a particular mutation will impair can be predicted, to some extent, by identifying the area of the AR in which the mutation resides. This predictive ability is primarily retrospective in origin; the different functional domains of the AR gene have been elucidated by analyzing the effects of specific mutations in different regions of the AR.[3] For example, mutations in the steroid binding domain have been known to affect androgen binding affinity or retention, mutations in the hinge region have been known to affect nuclear translocation, mutations in the DNA-binding domain have been known to affect dimerization and binding to target DNA, and mutations in the transactivation domain have been known to affect target gene transcription regulation.[3][81] Unfortunately, even when the affected functional domain is known, it is difficult to predict the phenotypical consequences of a particular mutation (see Correlation of genotype and phenotype).
Some mutations can adversely impact more than one functional domain. For example, a mutation in one functional domain can have deleterious effects on another by altering the way in which the domains interact.[81] A single mutation can affect all downstream functional domains if a premature stop codon or framing error results; such a mutation can result in a completely unusable (or unsynthesizable) androgen receptor protein.[3] The steroid binding domain is particularly vulnerable to the effects of a premature stop codon or framing error, since it occurs at the end of the gene, and its information is thus more likely to be truncated or misinterpreted than other functional domains.[3]
Other, more complex relationships have been observed as a consequence of mutated AR; some mutations associated with male phenotypes have been linked to male breast cancer, prostate cancer, or in the case of spinal and bulbar muscular atrophy, disease of the central nervous system.[9][24][88][89][90] The form of breast cancer that is seen in some men with partial androgen insensitivity syndrome is caused by a mutation in the AR's DNA-binding domain.[88][90] It has been hypothesized that this mutation causes a disturbance of the AR's target gene interaction that allows it to act at certain additional targets, possibly in conjunction with the estrogen receptor protein, to cause cancerous growth.[3] The etiology of spinal and bulbar muscular atrophy (SBMA) demonstrates that even the mutant AR protein itself can result in pathology. The trinucleotide repeat expansion of the polyglutamine tract of the AR gene that is associated with SBMA results in the synthesis of a misfolded AR protein that the cell fails to properly proteolyze and disperse.[91] These misfolded AR proteins form aggregates in the cell cytoplasm and nucleus.[91] Over the course of 30 to 50 years, these aggregates accumulate and have a cytotoxic effect, eventually resulting in the neurodegenerative symptoms associated with SBMA.[91]
Diagnosis
The phenotypes that result from the insensitivity to androgens are not unique to AIS, and thus the diagnosis of AIS requires thorough exclusion of other causes.[14][65] Clinical findings indicative of AIS include the presence of a short vagina [92] or undermasculinized genitalia,[1][64][83] partial or complete regression of Müllerian structures,[93] bilateral nondysplastic testes,[94] and impaired spermatogenesis and / or virilization.[1][5][6][9] Laboratory findings include a 46,XY karyotype [2] and normal or elevated postpubertal testosterone, luteinizing hormone, and estradiol levels.[2][14] The androgen binding activity of genital skin fibroblasts is typically diminished,[3][95] although exceptions have been reported.[96] Conversion of testosterone to dihydrotestosterone may be impaired.[3] The diagnosis of AIS is confirmed if androgen receptor gene sequencing reveals a mutation, although not all individuals with AIS (particularly PAIS) will have an AR mutation (see Other Causes).[2][6][48][49]
Each of the three types of AIS --- complete, partial, and mild --- has a different list of differential diagnoses to consider.[1] Depending on the form of AIS that is suspected, the list of differentials can include:[54][55][97][98][99]
- Chromosomal anomalies:
- Klinefelter syndrome (47,XXY karyotype)
- Turner syndrome (45,XO karyotype)
- Mixed gonadal dysgenesis (45,XO/46,XY karyotype)
- Tetragametic chimerism (46,XX/46,XY karyotype)
- Androgen biosynthetic dysfunction in 46,XY individuals:
- Luteinizing hormone (LH) receptor mutations
- Smith-Lemli-Opitz syndrome (associated with mental retardation)
- Lipoid congenital adrenal hyperplasia
- 3β-hydroxysteroid dehydrogenase 2 deficiency
- 17α-hydroxylase deficiency
- 17,20 lyase deficiency
- 17β-hydroxysteroid dehydrogenase deficiency
- 5α-reductase deficiency
- Androgen excess in 46,XX individuals:
- 21-hydroxylase deficiency
- 3β-hydroxysteroid dehydrogenase 2 deficiency
- Cytochrome P450 oxidoreductase deficiency (disorder in mother causes 46,XX fetal virilization)
- 11β-hydroxylase deficiency
- Aromatase deficiency
- Glucocorticoid receptor mutations
- Maternal virilizing tumor (e.g. luteoma)
- Increased androgen exposure in utero, not otherwise specified (e.g. androgenic drugs)
- Developmental
- Mayer-Rokitansky-Küster-Hauser syndrome (46,XX karyotype)
- Swyer syndrome (46,XY karyotype)
- XX gonadal dysgenesis (46,XX karyotype)
- Leydig cell agenesis or hypoplasia, not otherwise specified (46,XY karyotype)
- Absent (vanishing) testes syndrome
- Ovotesticular DSD
- Testicular DSD (i.e. 46,XX sex reversal)
- Teratogenic causes (e.g. estrogens, antiestrogens)
- Other causes:
- Frasier syndrome (associated with progressive glomerulopathy)
- Denys-Drash syndrome (associated with nephropathy and Wilms tumor)
- WAGR syndrome (associated with Wilms tumor and aniridia)
- McKusick-Kaufman syndrome (associated with postaxial polydactyly)
- Robinow syndrome (associated with dwarfism)
- Aarskog-Scott syndrome (associated with facial anomalies)
- Hand-foot-genital syndrome (associated with limb malformations)
- Popliteal pterygium syndrome (associated with extensive webbing behind knees)
- Kallmann syndrome (often associated with anosmia)
- Hypospadias not otherwise specified
- Cryptorchidism not otherwise specified
- vaginal atresia not otherwise specified
CAIS
Main article: Diagnosis of Complete Androgen Insensitivity SyndromePAIS
Main article: Diagnosis of Partial Androgen Insensitivity SyndromeMAIS
Main article: Diagnosis of Mild Androgen Insensitivity SyndromeManagement
Management of AIS is currently limited to symptomatic management; methods to correct a malfunctioning androgen receptor protein that result from an AR gene mutation are not currently available. Areas of management include sex assignment, genitoplasty, gonadectomy in relation to tumor risk, hormone replacement therapy, and genetic and psychological counseling.
CAIS
Main article: Management of Complete Androgen Insensitivity SyndromePAIS
Main article: Management of Partial Androgen Insensitivity SyndromeMAIS
Main article: Management of Mild Androgen Insensitivity SyndromeEpidemiology
Estimates for the incidence of androgen insensitivity syndrome are based on a relatively small population size, and thus are known to be imprecise.[1] CAIS is estimated to occur in 1 out of every 20,400 46,XY births.[100] A nationwide survey in The Netherlands based on patients with genetic confirmation of the diagnosis estimates that the minimal incidence of CAIS is 1 in 99,000.[63] The incidence of PAIS is estimated to be 1 in 130,000.[101] Due to its subtle presentation, MAIS is not typically investigated except in the case of male infertility,[83] and thus its true prevalence is unknown.[2]
History
Recorded descriptions of the effects of androgen insensitivity syndrome date back for hundreds of years, although significant understanding of its underlying histopathology would not occur until the 1950s.[1] The taxonomy and nomenclature associated with androgen insensitivity went through a significant evolution that paralleled this understanding.
Timeline of major milestones
- 1950: Lawson Wilkins administers daily methyltestosterone to a 46,XY female patient, who shows no signs of virilization. His experiment is the first documented demonstration of the pathophysiology of androgen insensitivity syndrome.[65][102]
- 1970: Mary F. Lyon and Susan Hawkes report that a gene on the X chromosome caused complete insensitivity to androgens in mice.[103][104]
- 1981: Barbara Migeon et al. narrow down the locus of the human androgen receptor gene (or a factor controlling the androgen receptor gene) to somewhere between Xq11 and Xq13.[105][106]
- 1988: The human androgen receptor gene is first cloned and partially analyzed by multiple parties.[107][108] Terry Brown et al. report the first mutations proven to cause AIS.[2][106]
- 1989: Terry Brown et al. reports the exact locus of the AR gene (Xq11-Xq12),[16] and Dennis Lubahn et al. publishes its intron-exon boundaries.[109]
- 1994: The androgen receptor gene mutations database is created to provide a comprehensive listing of mutations published in journals and conference proceedings.[110]
Early terminology
The first descriptions of the effects of androgen insensitivity appeared in the medical literature as individual case reports or as part of a comprehensive description of intersex physicalities. In 1839, Scottish obstetrician Sir James Young Simpson published one such description [111] in an exhaustive study of intersexuality that has been credited with advancing the medical community's understanding of the subject.[112] Simpson's system of taxonomy, however, was far from the first; taxonomies / descriptions for the classification of intersexuality were developed by Italian physician and physicist Fortuné Affaitati in 1549,[113][114] French surgeon Ambroise Paré in 1573,[112][115] French physician and sexology pioneer Nicolas Venette in 1687 (under the pseudonym Vénitien Salocini),[116][117] and French Zoologist Isidore Geoffroy St. Hilaire in 1832.[118] All five of the aforementioned authors used the colloquial term "hermaphrodite" as the foundation of their taxonomies, although Simpson himself questioned the propriety of the word in his publication.[111] Use of the word "hermaphrodite" in the medical literature has persisted to this day,[119][120] although its propriety is still in question. An alternative system of nomenclature has been recently suggested,[121] but the subject of exactly which word or words should be used in its place still one of much debate.[98][122][123][124][125]
 "Pudenda pseudo-hermaphroditi ovini." Illustration of ambiguous genitalia from Frederik Ruysch’s Thesaurus Anitomicus Octavius, 1709.[126]
"Pudenda pseudo-hermaphroditi ovini." Illustration of ambiguous genitalia from Frederik Ruysch’s Thesaurus Anitomicus Octavius, 1709.[126]Pseudohermaphroditism
"Pseudohermaphroditism" has, until very recently,[121] been the term used in the medical literature to describe the condition of an individual whose gonads and karyotype do not match the external genitalia in the gender binary sense. For example, 46,XY individuals that have a female phenotype, but also have testes instead of ovaries --- a group that includes all individuals with complete androgen insensitivity (CAIS), as well as some individuals with partial androgen insensitivity (PAIS) --- are classified as having "male pseudohermaphroditism," while individuals with both an ovary and a testis (or at least one ovotestis) are classified as having "true hermaphroditism.".[120][121] Usage of the word in the medical literature predates the discovery of the chromosome, and thus its definition has not always taken karyotype into account when determining an individual's sex. Previous definitions of "pseudohermaphroditism" relied on perceived inconsistencies between the internal and external organs; the "true" sex of an individual was determined by the internal organs, and the external organs determined the "perceived" sex of an individual.[111][118]
German-Swiss pathologist Edwin Klebs is sometimes noted for using the word "pseudohermaphroditism" in his taxonomy of intersexuality in 1876,[127] although the word is clearly not his invention as is sometimes reported; the history of the word "pseudohermaphrodite," and the corresponding desire to separate "true" hermaphrodites from "false," "spurious," or "pseudo" hermaphrodites, dates back to at least 1709, when Dutch anatomist Frederik Ruysch used it in a publication describing a subject with testes and a mostly female phenotype.[126] "Pseudohermaphrodite" also appeared in the Acta Eruditorum later that same year, in a review of Ruysch's work.[128] There is also evidence that the word was already being used by the German and French medical community long before Klebs used it; German physiologist Johannes Peter Müller equated "pseudohermaphroditism" with a sub-class of hermaphroditism from St. Hilaire's taxonomy in a publication dated 1834,[129] and by the 1840s "pseudo-hermaphroditism" was appearing in several French and German publications, including dictionaries.[130][131][132][133]
Testicular feminization
In 1953, American gynecologist John Morris provided the first full description of what he called "testicular feminization syndrome" based on 82 cases compiled from the medical literature, including 2 of his own patients.[1][3][134] The term "testicular feminization" was coined to reflect Morris' observation that the testicles in these patients produced a hormone that had a feminizing effect on the body, a phenomenon that is now understood to be due to the inaction of androgens, and subsequent aromatization of testosterone into estrogen.[1] A few years before Morris published his landmark paper, Lawson Wilkins had shown through his own experiments that unresponsiveness of the target cell to the action of androgenic hormones was a cause of "male pseudohermaphroditism".[65][102] Wilkins' work, which clearly demonstrated the lack of a therapeutic effect when 46,XY women were treated with androgens, caused a gradual shift in nomenclature from "testicular feminization" to "androgen resistance".[83]
Other names
A distinct name has been given to many of the various presentations of androgen insensitivity syndrome, such as Reifenstein syndrome (1947),[135] Goldberg-Maxwell syndrome (1948),[136] Morris' syndrome (1953),[134] Gilbert-Dreyfus syndrome (1957),[137] Lub's syndrome (1959),[138] "incomplete testicular feminization" (1963),[139] Rosewater syndrome (1965),[140] and Aiman's syndrome (1979).[141] Since it was not understood that these different presentations were all caused by the same set of mutations in the androgen receptor gene, a unique name was given to each new combination of symptoms, resulting in a complicated stratification of seemingly disparate disorders.[65][142]
Over the last 60 years, as reports of strikingly different phenotypes were reported to occur even among members of the same family, and as steady progress was made towards the understanding of the underlying molecular pathogenesis of AIS, it has been demonstrated that these disorders are different phenotypic expressions of one syndrome caused by molecular defects in the androgen receptor gene.[1][13][65][142]
Androgen insensitivity syndrome (AIS) is now the accepted terminology for the syndromes resulting from unresponsiveness of the target cell to the action of androgenic hormones.[1] AIS is broken down into three classes based on phenotype: complete androgen insensitivity syndrome (CAIS), partial androgen insensitivity syndrome (PAIS), and mild androgen insensitivity syndrome (MAIS).[1][2][5][6][7][10][11][12][13] CAIS encompasses the phenotypes previously described by "testicular feminization," Morris' syndrome, and Goldberg-Maxwell syndrome;[1][143] PAIS includes Reifenstein syndrome, Gilbert-Dreyfus syndrome, Lub's syndrome, "incomplete testicular feminization," and Rosewater syndrome;[142][144][145] and MAIS includes Aiman's syndrome.[146]
The more virilized phenotypes of AIS have sometimes been described as "undervirilized male syndrome," "infertile male syndrome," "undervirilized fertile male syndrome," etc., before evidence was reported that these conditions were caused by mutations in the androgen receptor gene.[59] These diagnoses were used to describe a variety of mild defects in virilization; as a result, the phenotypes of some men that have been diagnosed as such are better described by PAIS (e.g. micropenis, hypospadias and undescended testes), while others are better described by MAIS (e.g. isolated infertility or gynecomastia).[1][59][60][145][147][148]
Popular culture
The 1991 novel Ring (later adopted into English "The Ring" series) describes that the central antagonist Sadako) has this syndrome. In Season 2 Episode 13 "Skin Deep" of House
References
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab Hughes IA, Deeb A (December 2006). "Androgen resistance". Best Pract. Res. Clin. Endocrinol. Metab. 20 (4): 577–98. doi:10.1016/j.beem.2006.11.003. PMID 17161333.
- ^ a b c d e f g h i j k l m n o p Galani A, Kitsiou-Tzeli S, Sofokleous C, Kanavakis E, Kalpini-Mavrou A (2008). "Androgen insensitivity syndrome: clinical features and molecular defects". Hormones (Athens) 7 (3): 217–29. PMID 18694860.
- ^ a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab Quigley CA, De Bellis A, Marschke KB, el-Awady MK, Wilson EM, French FS (June 1995). "Androgen receptor defects: historical, clinical, and molecular perspectives". Endocr. Rev. 16 (3): 271–321. PMID 7671849.
- ^ a b c d e Giwercman YL, Nordenskjöld A, Ritzén EM, Nilsson KO, Ivarsson SA, Grandell U, Wedell A (June 2002). "An androgen receptor gene mutation (E653K) in a family with congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency as well as in partial androgen insensitivity". J. Clin. Endocrinol. Metab. 87 (6): 2623–8. doi:10.1210/jc.87.6.2623. PMID 12050225.
- ^ a b c d e Zuccarello D, Ferlin A, Vinanzi C, Prana E, Garolla A, Callewaert L, Claessens F, Brinkmann AO, Foresta C (April 2008). "Detailed functional studies on androgen receptor mild mutations demonstrate their association with male infertility". Clin. Endocrinol. (Oxf) 68 (4): 580–8. doi:10.1111/j.1365-2265.2007.03069.x. PMID 17970778.
- ^ a b c d e f g Ferlin A, Vinanzi C, Garolla A, Selice R, Zuccarello D, Cazzadore C, Foresta C (November 2006). "Male infertility and androgen receptor gene mutations: clinical features and identification of seven novel mutations". Clin. Endocrinol. (Oxf) 65 (5): 606–10. doi:10.1111/j.1365-2265.2006.02635.x. PMID 17054461.
- ^ a b c d Stouffs K, Tournaye H, Liebaers I, Lissens W (2009). "Male infertility and the involvement of the X chromosome". Hum. Reprod. Update 15 (6): 623–37. doi:10.1093/humupd/dmp023. PMID 19515807.
- ^ Giwercman YL, Nikoshkov A, Byström B, Pousette A, Arver S, Wedell A (June 2001). "A novel mutation (N233K) in the transactivating domain and the N756S mutation in the ligand binding domain of the androgen receptor gene are associated with male infertility". Clin. Endocrinol. (Oxf) 54 (6): 827–34. doi:10.1046/j.1365-2265.2001.01308.x. PMID 11422119.
- ^ a b c Lund A, Juvonen V, Lähdetie J, Aittomäki K, Tapanainen JS, Savontaus ML (June 2003). "A novel sequence variation in the transactivation regulating domain of the androgen receptor in two infertile Finnish men". Fertil. Steril.. 79 Suppl 3: 1647–8. PMID 12801573.
- ^ a b c d Ozülker T, Ozpaçaci T, Ozülker F, Ozekici U, Bilgiç R, Mert M (January 2010). "Incidental detection of Sertoli-Leydig cell tumor by FDG PET/CT imaging in a patient with androgen insensitivity syndrome". Ann Nucl Med 24 (1): 35–9. doi:10.1007/s12149-009-0321-x. PMID 19957213.
- ^ a b c d Davis-Dao CA, Tuazon ED, Sokol RZ, Cortessis VK (November 2007). "Male infertility and variation in CAG repeat length in the androgen receptor gene: a meta-analysis". J. Clin. Endocrinol. Metab. 92 (11): 4319–26. doi:10.1210/jc.2007-1110. PMID 17684052.
- ^ a b c Kawate H, Wu Y, Ohnaka K, Tao RH, Nakamura K, Okabe T, Yanase T, Nawata H, Takayanagi R (November 2005). "Impaired nuclear translocation, nuclear matrix targeting, and intranuclear mobility of mutant androgen receptors carrying amino acid substitutions in the deoxyribonucleic acid-binding domain derived from androgen insensitivity syndrome patients". J. Clin. Endocrinol. Metab. 90 (11): 6162–9. doi:10.1210/jc.2005-0179. PMID 16118342.
- ^ a b c d e f g h Gottlieb B, Lombroso R, Beitel LK, Trifiro MA (January 2005). "Molecular pathology of the androgen receptor in male (in)fertility". Reprod. Biomed. Online 10 (1): 42–8. doi:10.1016/S1472-6483(10)60802-4. PMID 15705293.
- ^ a b c Ahmed SF, Cheng A, Hughes IA (April 1999). "Assessment of the gonadotrophin-gonadal axis in androgen insensitivity syndrome". Arch. Dis. Child. 80 (4): 324–9. doi:10.1136/adc.80.4.324. PMC 1717906. PMID 10086936. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1717906.
- ^ Sultan C, Paris F, Terouanne B, Balaguer P, Georget V, Poujol N, Jeandel C, Lumbroso S, Nicolas JC (2001). "Disorders linked to insufficient androgen action in male children". Hum. Reprod. Update 7 (3): 314–22. doi:10.1093/humupd/7.3.314. PMID 11392378.
- ^ a b Brown CJ, Goss SJ, Lubahn DB, Joseph DR, Wilson EM, French FS, Willard HF (February 1989). "Androgen receptor locus on the human X chromosome: regional localization to Xq11-12 and description of a DNA polymorphism". Am. J. Hum. Genet. 44 (2): 264–9. PMC 1715398. PMID 2563196. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1715398.
- ^ a b Kooy RF, Reyniers E, Storm K, Vits L, van Velzen D, de Ruiter PE, Brinkmann AO, de Paepe A, Willems PJ (July 1999). "CAG repeat contraction in the androgen receptor gene in three brothers with mental retardation". Am. J. Med. Genet. 85 (3): 209–13. doi:10.1002/(SICI)1096-8628(19990730)85:3<209::AID-AJMG4>3.0.CO;2-2. PMID 10398229.
- ^ Dejager S, Bry-Gauillard H, Bruckert E, Eymard B, Salachas F, LeGuern E, Tardieu S, Chadarevian R, Giral P, Turpin G (August 2002). "A comprehensive endocrine description of Kennedy's disease revealing androgen insensitivity linked to CAG repeat length". J. Clin. Endocrinol. Metab. 87 (8): 3893–901. doi:10.1210/jc.87.8.3893. PMID 12161529.
- ^ a b c Choong CS, Wilson EM (December 1998). "Trinucleotide repeats in the human androgen receptor: a molecular basis for disease". J. Mol. Endocrinol. 21 (3): 235–57. doi:10.1677/jme.0.0210235. PMID 9845666.
- ^ Audi L, Fernández-Cancio M, Carrascosa A, et al. (April 2010). "Novel (60%) and recurrent (40%) androgen receptor gene mutations in a series of 59 patients with a 46,XY disorder of sex development". J. Clin. Endocrinol. Metab. 95 (4): 1876–88. doi:10.1210/jc.2009-2146. PMID 20150575.
- ^ Lumbroso R, Beitel LK, Vasiliou DM, Trifiro MA, Pinsky L (November 1997). "Codon-usage variants in the polymorphic (GGN)n trinucleotide repeat of the human androgen receptor gene". Hum. Genet. 101 (1): 43–6. doi:10.1007/s004390050583. PMID 9385367.
- ^ a b Gottlieb B, Pinsky L, Beitel LK, Trifiro M (December 1999). "Androgen insensitivity". Am. J. Med. Genet. 89 (4): 210–7. doi:10.1002/(SICI)1096-8628(19991229)89:4<210::AID-AJMG5>3.0.CO;2-P. PMID 10727996.
- ^ Edwards A, Hammond HA, Jin L, Caskey CT, Chakraborty R (February 1992). "Genetic variation at five trimeric and tetrameric tandem repeat loci in four human population groups". Genomics 12 (2): 241–53. doi:10.1016/0888-7543(92)90371-X. PMID 1740333.
- ^ a b Casella R, Maduro MR, Lipshultz LI, Lamb DJ (November 2001). "Significance of the polyglutamine tract polymorphism in the androgen receptor". Urology 58 (5): 651–6. doi:10.1016/S0090-4295(01)01401-7. PMID 11711330.
- ^ Yeh SH, Chiu CM, Chen CL, Lu SF, Hsu HC, Chen DS, Chen PJ (April 2007). "Somatic mutations at the trinucleotide repeats of androgen receptor gene in male hepatocellular carcinoma". Int. J. Cancer 120 (8): 1610–7. doi:10.1002/ijc.22479. PMID 17230529.
- ^ La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH (July 1991). "Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy". Nature 352 (6330): 77–9. doi:10.1038/352077a0. PMID 2062380.
- ^ Casella R, Maduro MR, Misfud A, Lipshultz LI, Yong EL, Lamb DJ (January 2003). "Androgen receptor gene polyglutamine length is associated with testicular histology in infertile patients". J. Urol. 169 (1): 224–7. doi:10.1097/01.ju.0000035361.18870.6e. PMID 12478141.
- ^ Dowsing AT, Yong EL, Clark M, McLachlan RI, de Kretser DM, Trounson AO (August 1999). "Linkage between male infertility and trinucleotide repeat expansion in the androgen-receptor gene". Lancet 354 (9179): 640–3. doi:10.1016/S0140-6736(98)08413-X. PMID 10466666.
- ^ Tut TG, Ghadessy FJ, Trifiro MA, Pinsky L, Yong EL (November 1997). "Long polyglutamine tracts in the androgen receptor are associated with reduced trans-activation, impaired sperm production, and male infertility". J. Clin. Endocrinol. Metab. 82 (11): 3777–82. doi:10.1210/jc.82.11.3777. PMID 9360540.
- ^ Lim HN, Chen H, McBride S, Dunning AM, Nixon RM, Hughes IA, Hawkins JR (March 2000). "Longer polyglutamine tracts in the androgen receptor are associated with moderate to severe undermasculinized genitalia in XY males". Hum. Mol. Genet. 9 (5): 829–34. doi:10.1093/hmg/9.5.829. PMID 10749991.
- ^ Hiort O, Holterhus PM, Horter T, Schulze W, Kremke B, Bals-Pratsch M, Sinnecker GH, Kruse K (August 2000). "Significance of mutations in the androgen receptor gene in males with idiopathic infertility". J. Clin. Endocrinol. Metab. 85 (8): 2810–5. doi:10.1210/jc.85.8.2810. PMID 10946887.
- ^ Kukuvitis A, Georgiou I, Bouba I, Tsirka A, Giannouli CH, Yapijakis C, Tarlatzis B, Bontis J, Lolis D, Sofikitis N, Papadimas J (June 2002). "Association of oestrogen receptor alpha polymorphisms and androgen receptor CAG trinucleotide repeats with male infertility: a study in 109 Greek infertile men". Int. J. Androl. 25 (3): 149–52. doi:10.1046/j.1365-2605.2002.00339.x. PMID 12031042.
- ^ von Eckardstein S, Syska A, Gromoll J, Kamischke A, Simoni M, Nieschlag E (June 2001). "Inverse correlation between sperm concentration and number of androgen receptor CAG repeats in normal men". J. Clin. Endocrinol. Metab. 86 (6): 2585–90. doi:10.1210/jc.86.6.2585. PMID 11397858.
- ^ Rajpert-De Meyts E, Leffers H, Petersen JH, Andersen AG, Carlsen E, Jørgensen N, Skakkebaek NE (January 2002). "CAG repeat length in androgen-receptor gene and reproductive variables in fertile and infertile men". Lancet 359 (9300): 44–6. doi:10.1016/S0140-6736(02)07280-X. PMID 11809188.
- ^ Hiort O, Horter T, Schulze W, Kremke B, Sinnecker GH (November 1999). "Male infertility and increased risk of diseases in future generations". Lancet 354 (9193): 1907–8. doi:10.1016/S0140-6736(05)76874-4. PMID 10584751.
- ^ Muroya K, Sasagawa I, Suzuki Y, Nakada T, Ishii T, Ogata T (May 2001). "Hypospadias and the androgen receptor gene: mutation screening and CAG repeat length analysis". Mol. Hum. Reprod. 7 (5): 409–13. doi:10.1093/molehr/7.5.409. PMID 11331662.
- ^ Radpour R, Rezaee M, Tavasoly A, Solati S, Saleki A (2007). "Association of long polyglycine tracts (GGN repeats) in exon 1 of the androgen receptor gene with cryptorchidism and penile hypospadias in Iranian patients". J. Androl. 28 (1): 164–9. doi:10.2164/jandrol.106.000927. PMID 16957138.
- ^ Aschim EL, Nordenskjöld A, Giwercman A, Lundin KB, Ruhayel Y, Haugen TB, Grotmol T, Giwercman YL (October 2004). "Linkage between cryptorchidism, hypospadias, and GGN repeat length in the androgen receptor gene". J. Clin. Endocrinol. Metab. 89 (10): 5105–9. doi:10.1210/jc.2004-0293. PMID 15472213.
- ^ Rajender S, Rajani V, Gupta NJ, Chakravarty B, Singh L, Thangaraj K (2006). "No association of androgen receptor GGN repeat length polymorphism with infertility in Indian men". J. Androl. 27 (6): 785–9. doi:10.2164/jandrol.106.000166. PMID 16809273.
- ^ a b Gottlieb B, Beitel LK, Trifiro MA (May 2001). "Variable expressivity and mutation databases: The androgen receptor gene mutations database". Hum. Mutat. 17 (5): 382–8. doi:10.1002/humu.1113. PMID 11317353.
- ^ a b Köhler B, Lumbroso S, Leger J, Audran F, Grau ES, Kurtz F, Pinto G, Salerno M, Semitcheva T, Czernichow P, Sultan C (January 2005). "Androgen insensitivity syndrome: somatic mosaicism of the androgen receptor in seven families and consequences for sex assignment and genetic counseling". J. Clin. Endocrinol. Metab. 90 (1): 106–11. doi:10.1210/jc.2004-0462. PMID 15522944.
- ^ Hiort O, Sinnecker GH, Holterhus PM, Nitsche EM, Kruse K (June 1998). "Inherited and de novo androgen receptor gene mutations: investigation of single-case families". J. Pediatr. 132 (6): 939–43. doi:10.1016/S0022-3476(98)70387-7. PMID 9627582.
- ^ Batch JA, Williams DM, Davies HR, Brown BD, Evans BA, Hughes IA, Patterson MN (October 1992). "Androgen receptor gene mutations identified by SSCP in fourteen subjects with androgen insensitivity syndrome". Hum. Mol. Genet. 1 (7): 497–503. doi:10.1093/hmg/1.7.497. PMID 1307250.
- ^ Hiort O, Klauber G, Cendron M, Sinnecker GH, Keim L, Schwinger E, Wolfe HJ, Yandell DW (May 1994). "Molecular characterization of the androgen receptor gene in boys with hypospadias". Eur. J. Pediatr. 153 (5): 317–21. doi:10.1007/BF01956409. PMID 8033918.
- ^ Lu J, Danielsen M (June 1996). "A Stu I polymorphism in the human androgen receptor gene (AR)". Clin. Genet. 49 (6): 323–4. doi:10.1111/j.1399-0004.1996.tb03800.x. PMID 8884086.
- ^ Macke JP, Hu N, Hu S, Bailey M, King VL, Brown T, Hamer D, Nathans J (October 1993). "Sequence variation in the androgen receptor gene is not a common determinant of male sexual orientation". Am. J. Hum. Genet. 53 (4): 844–52. PMC 1682384. PMID 8213813. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1682384.
- ^ a b Gottlieb B, Vasiliou DM, Lumbroso R, Beitel LK, Pinsky L, Trifiro MA (1999). "Analysis of exon 1 mutations in the androgen receptor gene". Hum. Mutat. 14 (6): 527–39. doi:10.1002/(SICI)1098-1004(199912)14:6<527::AID-HUMU12>3.0.CO;2-X. PMID 10571951.
- ^ a b Melo KF, Mendonca BB, Billerbeck AE, Costa EM, Inácio M, Silva FA, Leal AM, Latronico AC, Arnhold IJ (July 2003). "Clinical, hormonal, behavioral, and genetic characteristics of androgen insensitivity syndrome in a Brazilian cohort: five novel mutations in the androgen receptor gene". J. Clin. Endocrinol. Metab. 88 (7): 3241–50. doi:10.1210/jc.2002-021658. PMID 12843171.
- ^ a b Ahmed SF, Cheng A, Dovey L, Hawkins JR, Martin H, Rowland J, Shimura N, Tait AD, Hughes IA (February 2000). "Phenotypic features, androgen receptor binding, and mutational analysis in 278 clinical cases reported as androgen insensitivity syndrome". J. Clin. Endocrinol. Metab. 85 (2): 658–65. doi:10.1210/jc.85.2.658. PMID 10690872.
- ^ a b Coutant R, Mallet D, Lahlou N, Bouhours-Nouet N, Guichet A, Coupris L, Croué A, Morel Y (August 2007). "Heterozygous mutation of steroidogenic factor-1 in 46,XY subjects may mimic partial androgen insensitivity syndrome". J. Clin. Endocrinol. Metab. 92 (8): 2868–73. doi:10.1210/jc.2007-0024. PMID 17488792.
- ^ a b c Adachi M, Takayanagi R, Tomura A, Imasaki K, Kato S, Goto K, Yanase T, Ikuyama S, Nawata H (September 2000). "Androgen-insensitivity syndrome as a possible coactivator disease". N. Engl. J. Med. 343 (12): 856–62. doi:10.1056/NEJM200009213431205. PMID 10995865.
- ^ Ghadessy FJ, Lim J, Abdullah AA, Panet-Raymond V, Choo CK, Lumbroso R, Tut TG, Gottlieb B, Pinsky L, Trifiro MA, Yong EL (June 1999). "Oligospermic infertility associated with an androgen receptor mutation that disrupts interdomain and coactivator (TIF2) interactions". J. Clin. Invest. 103 (11): 1517–25. doi:10.1172/JCI4289. PMC 408364. PMID 10359561. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=408364.
- ^ a b c d Giwercman YL, Ivarsson SA, Richthoff J, Lundin KB, Giwercman A (2004). "A novel mutation in the D-box of the androgen receptor gene (S597R) in two unrelated individuals Is associated with both normal phenotype and severe PAIS". Horm. Res. 61 (2): 58–62. doi:10.1159/000075240. PMID 14646391.
- ^ a b c d Achermann JC, Jameson JL (2006). "Disorders of sexual differentiation". In Hauser SL, Kasper DL, Fauci AS, Braunwald E, Longo DL. Harrison's endocrinology. New York: McGraw-Hill Medical Pub. Division. pp. 161–172. ISBN 0-07-145744-5.
- ^ a b c d Simpson JL, Rebar RW (2002). Hung, Wellington; Becker, Kenneth L.; Bilezikian, John P.; William J Bremner. ed. Principles and Practice of Endocrinology and Metabolism. Hagerstwon, MD: Lippincott Williams & Wilkins. pp. 852–885. ISBN 0-7817-4245-5.
- ^ Brinkmann A, Jenster G, Ris-Stalpers C, van der Korput H, Brüggenwirth H, Boehmer A, Trapman J (April 1996). "Molecular basis of androgen insensitivity". Steroids 61 (4): 172–5. doi:10.1016/0039-128X(96)00008-6. PMID 8732995.
- ^ Pinsky L, Kaufman M, Killinger DW (January 1989). "Impaired spermatogenesis is not an obligate expression of receptor-defective androgen resistance". Am. J. Med. Genet. 32 (1): 100–4. doi:10.1002/ajmg.1320320121. PMID 2705470.
- ^ Grino PB, Griffin JE, Cushard WG, Wilson JD (April 1988). "A mutation of the androgen receptor associated with partial androgen resistance, familial gynecomastia, and fertility". J. Clin. Endocrinol. Metab. 66 (4): 754–61. doi:10.1210/jcem-66-4-754. PMID 3346354.
- ^ a b c Tsukada T, Inoue M, Tachibana S, Nakai Y, Takebe H (October 1994). "An androgen receptor mutation causing androgen resistance in undervirilized male syndrome". J. Clin. Endocrinol. Metab. 79 (4): 1202–7. doi:10.1210/jc.79.4.1202. PMID 7962294.
- ^ a b Giwercman A, Kledal T, Schwartz M, Giwercman YL, Leffers H, Zazzi H, Wedell A, Skakkebaek NE (June 2000). "Preserved male fertility despite decreased androgen sensitivity caused by a mutation in the ligand-binding domain of the androgen receptor gene". J. Clin. Endocrinol. Metab. 85 (6): 2253–9. doi:10.1210/jc.85.6.2253. PMID 10852459.
- ^ Yong EL, Ng SC, Roy AC, Yun G, Ratnam SS (September 1994). "Pregnancy after hormonal correction of severe spermatogenic defect due to mutation in androgen receptor gene". Lancet 344 (8925): 826–7. doi:10.1016/S0140-6736(94)92385-X. PMID 7993455.
- ^ Bouvattier C, Mignot B, Lefèvre H, Morel Y, Bougnères P (September 2006). "Impaired sexual activity in male adults with partial androgen insensitivity". J. Clin. Endocrinol. Metab. 91 (9): 3310–5. doi:10.1210/jc.2006-0218. PMID 16757528.
- ^ a b c Boehmer AL, Brinkmann O, Brüggenwirth H, van Assendelft C, Otten BJ, Verleun-Mooijman MC, Niermeijer MF, Brunner HG, Rouwé CW, Waelkens JJ, Oostdijk W, Kleijer WJ, van der Kwast TH, de Vroede MA, Drop SL (September 2001). "Genotype versus phenotype in families with androgen insensitivity syndrome". J. Clin. Endocrinol. Metab. 86 (9): 4151–60. doi:10.1210/jc.86.9.4151. PMID 11549642.
- ^ a b Evans BA, Hughes IA, Bevan CL, Patterson MN, Gregory JW (June 1997). "Phenotypic diversity in siblings with partial androgen insensitivity syndrome". Arch. Dis. Child. 76 (6): 529–31. doi:10.1136/adc.76.6.529. PMC 1717223. PMID 9245853. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1717223.
- ^ a b c d e f Pérez-Palacios G, Chávez B, Méndez JP, McGinley JI, Ulloa-Aguirre A (1987). "The syndromes of androgen resistance revisited". J. Steroid Biochem. 27 (4–6): 1101–8. doi:10.1016/0022-4731(87)90196-8. PMID 3320547.
- ^ Radmayr C, Culig Z, Glatzl J, Neuschmid-Kaspar F, Bartsch G, Klocker H (October 1997). "Androgen receptor point mutations as the underlying molecular defect in 2 patients with androgen insensitivity syndrome". J. Urol. 158 (4): 1553–6. doi:10.1016/S0022-5347(01)64279-4. PMID 9302173.
- ^ Deeb A, Mason C, Lee YS, Hughes IA (July 2005). "Correlation between genotype, phenotype and sex of rearing in 111 patients with partial androgen insensitivity syndrome". Clin. Endocrinol. (Oxf) 63 (1): 56–62. doi:10.1111/j.1365-2265.2005.02298.x. PMID 15963062.
- ^ Rodien P, Mebarki F, Mowszowicz I, Chaussain JL, Young J, Morel Y, Schaison G (August 1996). "Different phenotypes in a family with androgen insensitivity caused by the same M780I point mutation in the androgen receptor gene". J. Clin. Endocrinol. Metab. 81 (8): 2994–8. doi:10.1210/jc.81.8.2994. PMID 8768864.
- ^ Nordenskjöld A, Söderhäll S (1998). "An androgen receptor gene mutation (A645D) in a boy with a normal phenotype". Hum. Mutat. 11 (4): 339. PMID 9554755.
- ^ Werner R, Holterhus PM, Binder G, Schwarz HP, Morlot M, Struve D, Marschke C, Hiort O (September 2006). "The A645D mutation in the hinge region of the human androgen receptor (AR) gene modulates AR activity, depending on the context of the polymorphic glutamine and glycine repeats". J. Clin. Endocrinol. Metab. 91 (9): 3515–20. doi:10.1210/jc.2006-0372. PMID 16804045.
- ^ Zenteno JC, Chávez B, Vilchis F, Kofman-Alfaro S (2002). "Phenotypic heterogeneity associated with identical mutations in residue 870 of the androgen receptor". Horm. Res. 57 (3–4): 90–3. doi:10.1159/000057958. PMID 12006704.
- ^ Holterhus PM, Werner R, Hoppe U, Bassler J, Korsch E, Ranke MB, Dörr HG, Hiort O. Molecular features and clinical phenotypes in androgen insensitivity syndrome in the absence and presence of androgen receptor gene mutations. J Mol Med. 2005;83:1005-1113.
- ^ Meehan KL, Sadar MD (May 2003). "Androgens and androgen receptor in prostate and ovarian malignancies". Front. Biosci. 8 (1–3): d780–800. doi:10.2741/1063. PMID 12700055.
- ^ a b Wang Q, Ghadessy FJ, Trounson A, de Kretser D, McLachlan R, Ng SC, Yong EL (December 1998). "Azoospermia associated with a mutation in the ligand-binding domain of an androgen receptor displaying normal ligand binding, but defective trans-activation". J. Clin. Endocrinol. Metab. 83 (12): 4303–9. doi:10.1210/jc.83.12.4303. PMID 9851768.
- ^ Taneja SS, Ha S, Swenson NK, Huang HY, Lee P, Melamed J, Shapiro E, Garabedian MJ, Logan SK (December 2005). "Cell-specific regulation of androgen receptor phosphorylation in vivo". J. Biol. Chem. 280 (49): 40916–24. doi:10.1074/jbc.M508442200. PMID 16210317.
- ^ Heinlein CA, Chang C (April 2002). "Androgen receptor (AR) coregulators: an overview". Endocr. Rev. 23 (2): 175–200. doi:10.1210/er.23.2.175. PMID 11943742.
- ^ Jenster G, van der Korput HA, van Vroonhoven C, van der Kwast TH, Trapman J, Brinkmann AO (October 1991). "Domains of the human androgen receptor involved in steroid binding, transcriptional activation, and subcellular localization". Mol. Endocrinol. 5 (10): 1396–404. doi:10.1210/mend-5-10-1396. PMID 1775129.
- ^ Simental JA, Sar M, Lane MV, French FS, Wilson EM (January 1991). "Transcriptional activation and nuclear targeting signals of the human androgen receptor". J. Biol. Chem. 266 (1): 510–8. PMID 1985913.
- ^ a b Gilbert SF (2000). Developmental biology. Sunderland, Mass: Sinauer Associates. ISBN 0-87893-243-7.
- ^ a b c d e f Jones RE, Lopez KH (2006). "Chapter 5: Sexual differentiation". Human reproductive biology. Amsterdam: Elsevier Academic Press. pp. 127–148. ISBN 0-12-088465-8.
- ^ a b c Yong EL, Loy CJ, Sim KS (2003). "Androgen receptor gene and male infertility". Hum. Reprod. Update 9 (1): 1–7. doi:10.1093/humupd/dmg003. PMID 12638777.
- ^ Hannema SE, Scott IS, Hodapp J, Martin H, Coleman N, Schwabe JW, Hughes IA (November 2004). "Residual activity of mutant androgen receptors explains wolffian duct development in the complete androgen insensitivity syndrome". J. Clin. Endocrinol. Metab. 89 (11): 5815–22. doi:10.1210/jc.2004-0709. PMID 15531547.
- ^ a b c d e Oakes MB, Eyvazzadeh AD, Quint E, Smith YR (December 2008). "Complete androgen insensitivity syndrome--a review". J Pediatr Adolesc Gynecol 21 (6): 305–10. doi:10.1016/j.jpag.2007.09.006. PMID 19064222.
- ^ a b Roy AK, Lavrovsky Y, Song CS, Chen S, Jung MH, Velu NK, Bi BY, Chatterjee B (1999). "Regulation of androgen action". Vitam. Horm.. Vitamins & Hormones 55: 309–52. doi:10.1016/S0083-6729(08)60938-3. ISBN 9780127098555. PMID 9949684.
- ^ a b Kokontis JM, Liao S (1999). "Molecular action of androgen in the normal and neoplastic prostate". Vitam. Horm. 55: 219–307. PMID 9949683.
- ^ a b Rajender S, Gupta NJ, Chakrabarty B, Singh L, Thangaraj K (March 2009). "Ala 586 Asp mutation in androgen receptor disrupts transactivation function without affecting androgen binding". Fertil. Steril. 91 (3): 933.e23–8. doi:10.1016/j.fertnstert.2008.10.041. PMID 19062009.
- ^ a b Sobel V, Schwartz B, Zhu YS, Cordero JJ, Imperato-McGinley J (August 2006). "Bone mineral density in the complete androgen insensitivity and 5alpha-reductase-2 deficiency syndromes". J. Clin. Endocrinol. Metab. 91 (8): 3017–23. doi:10.1210/jc.2005-2809. PMID 16735493.
- ^ a b Wooster R, Mangion J, Eeles R, Smith S, Dowsett M, Averill D, Barrett-Lee P, Easton DF, Ponder BA, Stratton MR (October 1992). "A germline mutation in the androgen receptor gene in two brothers with breast cancer and Reifenstein syndrome". Nat. Genet. 2 (2): 132–4. doi:10.1038/ng1092-132. PMID 1303262.
- ^ Evans BA, Harper ME, Daniells CE, Watts CE, Matenhelia S, Green J, Griffiths K (March 1996). "Low incidence of androgen receptor gene mutations in human prostatic tumors using single strand conformation polymorphism analysis". Prostate 28 (3): 162–71. doi:10.1002/(SICI)1097-0045(199603)28:3<162::AID-PROS3>3.0.CO;2-H. PMID 8628719.
- ^ a b Lobaccaro JM, Lumbroso S, Belon C, Galtier-Dereure F, Bringer J, Lesimple T, Namer M, Cutuli BF, Pujol H, Sultan C (November 1993). "Androgen receptor gene mutation in male breast cancer". Hum. Mol. Genet. 2 (11): 1799–802. doi:10.1093/hmg/2.11.1799. PMID 8281139.
- ^ a b c Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, Marcelli M, Weigel NL, Mancini MA (May 1999). "Polyglutamine-expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components and SRC-1, and are suppressed by the HDJ-2 chaperone". Hum. Mol. Genet. 8 (5): 731–41. doi:10.1093/hmg/8.5.731. PMID 10196362.
- ^ Ismail-Pratt IS, Bikoo M, Liao LM, Conway GS, Creighton SM (July 2007). "Normalization of the vagina by dilator treatment alone in Complete Androgen Insensitivity Syndrome and Mayer-Rokitansky-Kuster-Hauser Syndrome". Hum. Reprod. 22 (7): 2020–4. doi:10.1093/humrep/dem074. PMID 17449508.
- ^ Nichols JL, Bieber EJ, Gell JS. Case of sisters with complete androgen insensitivity syndrome and discordant Müllerian remnants. Fertil Steril. 2009;91:932e15-e18.
- ^ Hannema SE, Scott IS, Rajpert-De Meyts E, Skakkebaek NE, Coleman N, Hughes IA (March 2006). "Testicular development in the complete androgen insensitivity syndrome". J. Pathol. 208 (4): 518–27. doi:10.1002/path.1890. PMID 16400621.
- ^ Weidemann W, Linck B, Haupt H, Mentrup B, Romalo G, Stockklauser K, Brinkmann AO, Schweikert HU, Spindler KD (December 1996). "Clinical and biochemical investigations and molecular analysis of subjects with mutations in the androgen receptor gene". Clin. Endocrinol. (Oxf) 45 (6): 733–9. doi:10.1046/j.1365-2265.1996.8600869.x. PMID 9039340.
- ^ Deeb A, Jääskeläinen J, Dattani M, Whitaker HC, Costigan C, Hughes IA (October 2008). "A novel mutation in the human androgen receptor suggests a regulatory role for the hinge region in amino-terminal and carboxy-terminal interactions". J. Clin. Endocrinol. Metab. 93 (10): 3691–6. doi:10.1210/jc.2008-0737. PMID 18697867.
- ^ Quint EH, McCarthy JD, Smith YR (March 2010). "Vaginal surgery for congenital anomalies". Clin Obstet Gynecol 53 (1): 115–24. doi:10.1097/GRF.0b013e3181cd4128. PMID 20142648.
- ^ a b Hughes IA (February 2008). "Disorders of sex development: a new definition and classification". Best Pract. Res. Clin. Endocrinol. Metab. 22 (1): 119–34. doi:10.1016/j.beem.2007.11.001. PMID 18279784.
- ^ Kim KR, Kwon Y, Joung JY, Kim KS, Ayala AG, Ro JY (October 2002). "True hermaphroditism and mixed gonadal dysgenesis in young children: a clinicopathologic study of 10 cases". Mod. Pathol. 15 (10): 1013–9. doi:10.1097/01.MP.0000027623.23885.0D. PMID 12379746.
- ^ Bangsbøll S, Qvist I, Lebech PE, Lewinsky M (January 1992). "Testicular feminization syndrome and associated gonadal tumors in Denmark". Acta Obstet Gynecol Scand 71 (1): 63–6. doi:10.3109/00016349209007950. PMID 1315102.
- ^ Mazen I, El-Ruby M, Kamal R, El-Nekhely I, El-Ghandour M, Tantawy S, El-Gammal M (2010). "Screening of genital anomalies in newborns and infants in two egyptian governorates". Horm Res Paediatr 73 (6): 438–42. doi:10.1159/000313588. PMID 20407231.
- ^ a b Wilkins L. Heterosexual development. In: The diagnosis and treatment of endocrine disorders in childhood and adolescence. Springfield, IL: Charles C Thomas, 1950, pp. 256-279.
- ^ Lyon MF, Hawkes SG (September 1970). "X-linked gene for testicular feminization in the mouse". Nature 227 (5264): 1217–9. doi:10.1038/2271217a0. PMID 5452809.
- ^ Ohno S, Lyon MF (July 1970). "X-Linked testicular feminization in the mouse as a non-inducible regulatory mutation of the Jacob-Monod type". Clinical Genetics 1 (3–4): 121–127. doi:10.1111/j.1399-0004.1970.tb01627.x.
- ^ Migeon BR, Brown TR, Axelman J, Migeon CJ (October 1981). "Studies of the locus for androgen receptor: localization on the human X chromosome and evidence for homology with the Tfm locus in the mouse". Proc. Natl. Acad. Sci. U.S.A. 78 (10): 6339–43. doi:10.1073/pnas.78.10.6339. PMC 349034. PMID 6947233. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=349034.
- ^ a b Brown TR, Lubahn DB, Wilson EM, Joseph DR, French FS, Migeon CJ (November 1988). "Deletion of the steroid-binding domain of the human androgen receptor gene in one family with complete androgen insensitivity syndrome: evidence for further genetic heterogeneity in this syndrome". Proc. Natl. Acad. Sci. U.S.A. 85 (21): 8151–5. doi:10.1073/pnas.85.21.8151. PMC 282385. PMID 3186717. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=282385.
- ^ Lubahn DB, Joseph DR, Sullivan PM, Willard HF, French FS, Wilson EM (April 1988). "Cloning of human androgen receptor complementary DNA and localization to the X chromosome". Science 240 (4850): 327–30. doi:10.1126/science.3353727. PMID 3353727.
- ^ Chang CS, Kokontis J, Liao ST (April 1988). "Molecular cloning of human and rat complementary DNA encoding androgen receptors". Science 240 (4850): 324–6. doi:10.1126/science.3353726. PMID 3353726.
- ^ Lubahn DB, Brown TR, Simental JA, Higgs HN, Migeon CJ, Wilson EM, French FS (December 1989). "Sequence of the intron/exon junctions of the coding region of the human androgen receptor gene and identification of a point mutation in a family with complete androgen insensitivity". Proc. Natl. Acad. Sci. U.S.A. 86 (23): 9534–8. doi:10.1073/pnas.86.23.9534. PMC 298531. PMID 2594783. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=298531.
- ^ Patterson MN, Hughes IA, Gottlieb B, Pinsky L (September 1994). "The androgen receptor gene mutations database". Nucleic Acids Res. 22 (17): 3560–2. PMC 308319. PMID 7937057. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=308319.
- ^ a b c Simpson JY. Hermaphroditism. In: Todd RB, ed. Cyclopaedia of Anatomy and Physiology, Vol II. London: Longman, Brown, Green, Longmans, & Roberts 1839;2:684-738.
- ^ a b King H (2007). Midwifery, obstetrics and the rise of gynaecology: the uses of a sixteenth-century compendium. Aldershot, Hants, England: Ashgate Pub. ISBN 0-7546-5396-X.
- ^ Affaitati F [Affaitat]. De hermaphroditis. Venet. 1549.
- ^ Panckoucke CLF, ed. Dictionnaire des sciences médicales - biographie médicale, 1st ed. Paris: Panckoucke 1820;1:59.
- ^ Paré, A. Des monstres et prodiges. Paris: Dupuys 1573.
- ^ Venette N [Vénitien Salocini]. Tableau de l'amour humain considéré dans l'état du mariage. A Parme: Chez Franc d'Amour 1687.
- ^ Jacob G. Tractatus de hermaphroditis. London: E. Curll 1718.
- ^ a b Saint Hilaire IG. Histoire générale et particulière des anomalies de l'organisation. Paris: J.-B. Baillière 1832-1836.
- ^ Dorsey FY, Hsieh MH, Roth DR (March 2009). "46,XX SRY-negative true hermaphrodite siblings". Urology 73 (3): 529–31. doi:10.1016/j.urology.2008.09.050. PMID 19038427.
- ^ a b Verkauskas G, Jaubert F, Lortat-Jacob S, Malan V, Thibaud E, Nihoul-Fékété C (February 2007). "The long-term followup of 33 cases of true hermaphroditism: a 40-year experience with conservative gonadal surgery". J. Urol. 177 (2): 726–31; discussion 731. doi:10.1016/j.juro.2006.10.003. PMID 17222668.
- ^ a b c Hughes IA, Houk C, Ahmed SF, Lee PA (July 2006). "Consensus statement on management of intersex disorders". Arch. Dis. Child. 91 (7): 554–63. doi:10.1136/adc.2006.098319. PMC 2082839. PMID 16624884. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2082839.
- ^ Simmonds M (January 2007). "Was "variations of reproductive development" considered?". Arch. Dis. Child. 92 (1): 89. doi:10.1136/adc.2006.107797. PMC 2083124. PMID 17185456. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2083124.
- ^ Zannoni GF, Vellone VG, Cordisco EL, Sangiorgi E, Grimaldi ME, Neri C, Nanni L, Neri G (January 2010). "Morphology and immunophenotyping of a monolateral ovotestis in a 46,XderY/45,X mosaic individual with ambiguous genitalia". Int. J. Gynecol. Pathol. 29 (1): 33–8. doi:10.1097/PGP.0b013e3181b52e75. PMID 19952940.
- ^ Feder EK, Karkazis K (2008). "What's in a name? The controversy over "disorders of sex development"". Hastings Cent Rep 38 (5): 33–6. doi:10.1353/hcr.0.0062. PMID 18947138.
- ^ Reis E (2007). "Divergence or disorder?: the politics of naming intersex". Perspect. Biol. Med. 50 (4): 535–43. doi:10.1353/pbm.2007.0054. PMID 17951887.
- ^ a b Ruysch F. Thesaurus anatomicus octavus. Amsterdam: Joannem Wolters 1709. p. 33, Plate II.
- ^ Klebs E. Handbuch der pathologischen anatomie. Berlin: A. Hirschwald 1876;1:718.
- ^ Mencke JB, ed. Acta eruditorum anno mdccix. Leipzig: Joh. Grossii Haeredes, Joh. Frid. Gleditsch, & Frid. Groschuf. 1709;28:272-274.
- ^ Müller JP, ed. Archiv für Anatomie, Physiologie und wissenschaftliche Medicin. Berlin: G. Eichler 1834, p. 171.
- ^ Académie française. Complément du Dictionnaire de l'Académie française. Paris: Chez Firmin Didot Fréres 1843, p. 997.
- ^ Ritter von Raiman JN, Edlen von Rosas A, Fischer SC, Wisgrill J, eds. Medicinische Jahrbücher des kaiserlich-königlichen österreichischen Staates (volume 22). Vienna: Carl Gerold 1840;22:380-384.
- ^ Bertuch FJ, Schütz CG, eds. Allgemeine Literatur-Zeitung Issues 1-97. Leipzig 1815, pp. 257-260.
- ^ Peschier A, Mozin DJ, eds. Supplément au dictionnaire complet des langues française et allemande de l'abbe Mozin. Paris: Stuttgart et Augsbourg 1859, p. 333.
- ^ a b Morris JM (June 1953). "The syndrome of testicular feminization in male pseudohermaphrodites". Am. J. Obstet. Gynecol. 65 (6): 1192–1211. PMID 13057950.
- ^ Reifenstein EC Jr. (1947). "Hereditary familial hypogonadism". Proc Am Fed Clin Res 3: 86. PMID 18909356.
- ^ oldberg MB, Maxwell A (May 1948). "Male pseudohermaphroditism proved by surgical exploration and microscopic examination; a case report with speculations concerning pathogenesis". J. Clin. Endocrinol. Metab. 8 (5): 367–79. doi:10.1210/jcem-8-5-367. PMID 18863968.
- ^ Gilbert-Dreyfus S, Sabaoun CIA, Belausch J. Etude d'un cas familial d'androgynoidisme avec hypospadias grave, gynecomastie et hyperoestrogenie. Ann Endocr. 1957;18:93-101.
- ^ ubs HA Jr, Vilar O, Bergenstal DM (September 1959). "Familial male pseudohermaphrodism with labial testes and partial feminization: endocrine studies and genetic aspects". J. Clin. Endocrinol. Metab. 19 (9): 1110–20. doi:10.1210/jcem-19-9-1110. PMID 14418653.
- ^ Morris JM, Mahesh VB (November 1963). "Further observations on the syndrome, "testicular feminization."". Am. J. Obstet. Gynecol. 87: 731–48. PMID 14085776.
- ^ Rosewater S, Gwinup G, Hamwi JG (September 1965). "Familial gynecomastia". Ann. Intern. Med. 63: 377–85. PMID 14327504.
- ^ Aiman J, Griffin JE, Gazak JM, Wilson JD, MacDonald PC (February 1979). "Androgen insensitivity as a cause of infertility in otherwise normal men". N. Engl. J. Med. 300 (5): 223–7. doi:10.1056/NEJM197902013000503. PMID 759869.
- ^ a b c Simpson JL (2008). "Male Pseudohermaphroditism Due to Androgen Insensitivity or 5α-Reductase Deficiency". Glob. Libr. Women's Med.. doi:10.3843/GLOWM.10349.
- ^ Hester JD. Intersex(e) und alternative Heilungsstrategien - Medizin, soziale Imperative und identitatsstiftende Gegengemeinschaften. Ethik Med. 2004;16:48-67.
- ^ McPhaul MJ (1999). "Molecular defects of the androgen receptor". J. Steroid Biochem. Mol. Biol. 69 (1–6): 315–22. doi:10.1016/S0960-0760(99)00050-3. PMID 10419008.
- ^ a b Hoff, TA, Fuqua, SAW (2000). "Steroid and nuclear receptor polymorphism variants in hormone resistance and hormone independence". In Miller MS, Cronin MT. Genetic polymorphisms and susceptibility to disease. Washington, DC: Taylor & Francis. p. 111. ISBN 0-7484-0822-3.
- ^ Sultan C, Lumbroso S, Paris F, Jeandel C, Terouanne B, Belon C, Audran F, Poujol N, Georget V, Gobinet J, Jalaguier S, Auzou G, Nicolas JC (August 2002). "Disorders of androgen action". Semin. Reprod. Med. 20 (3): 217–28. doi:10.1055/s-2002-35386. PMID 12428202.
- ^ Chu J, Zhang R, Zhao Z, Zou W, Han Y, Qi Q, Zhang H, Wang JC, Tao S, Liu X, Luo Z (January 2002). "Male fertility is compatible with an Arg(840)Cys substitution in the AR in a large Chinese family affected with divergent phenotypes of AR insensitivity syndrome". J. Clin. Endocrinol. Metab. 87 (1): 347–51. doi:10.1210/jc.87.1.347. PMID 11788673.
- ^ Meschede D, Horst J (May 1997). "The molecular genetics of male infertility". Mol. Hum. Reprod. 3 (5): 419–30. doi:10.1093/molehr/3.5.419. PMID 9239727.
External links
Information
- Androgen Insensitivity Syndrome at NIH/UW GeneTests
- Online 'Mendelian Inheritance in Man' (OMIM) Androgen Insensitivity Syndrome -300068,313700
- An Australian parent/patient booklet on CAIS
- The Secret of My Sex news article
- Women With Male DNA All Female news article at ABCnews.com
Patient groups
- AIS Support Group AISSG (UK and International)
- AIS-DSD Support Group for Women & Families
- AIS Support Group (Australasia)
- Intersex Support Forums (US and International)
Sex linkage: X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) · Wiskott–Aldrich syndrome · X-linked severe combined immunodeficiency · X-linked agammaglobulinemia · Hyper-IgM syndrome type 1 · IPEX · X-linked lymphoproliferative disease · Properdin deficiencyHematologic Endocrine Androgen insensitivity syndrome/Kennedy disease · KAL1 Kallmann syndrome · X-linked adrenal hypoplasia congenitaMetabolic amino acid: Ornithine transcarbamylase deficiency · Oculocerebrorenal syndrome
dyslipidemia: Adrenoleukodystrophy
carbohydrate metabolism: Glucose-6-phosphate dehydrogenase deficiency · Pyruvate dehydrogenase deficiency · Danon disease/glycogen storage disease Type IIb
lipid storage disorder: Fabry's disease
mucopolysaccharidosis: Hunter syndrome
purine-pyrimidine metabolism: Lesch–Nyhan syndrome
mineral: Menkes disease/Occipital horn syndromeNervous system X-Linked mental retardation: Coffin–Lowry syndrome · MASA syndrome · X-linked alpha thalassemia mental retardation syndrome · Siderius X-linked mental retardation syndrome
eye disorders: Color blindness (red and green, but not blue) · Ocular albinism (1) · Norrie disease · Choroideremia
other: Charcot–Marie–Tooth disease (CMTX2-3) · Pelizaeus–Merzbacher disease · SMAX2Skin and related tissue Dyskeratosis congenita · Hypohidrotic ectodermal dysplasia (EDA) ·
X-linked ichthyosis · X-linked endothelial corneal dystrophyNeuromuscular Urologic Bone/tooth No primary system Barth syndrome · McLeod syndrome · Smith-Fineman-Myers syndrome · Simpson–Golabi–Behmel syndrome · Mohr–Tranebjærg syndrome · Nasodigitoacoustic syndromeX-linked dominant X-linked hypophosphatemia · Focal dermal hypoplasia · Fragile X syndrome · Aicardi syndrome · Incontinentia pigmenti · Rett syndrome · CHILD syndrome · Lujan–Fryns syndrome · Orofaciodigital syndrome 1Genetic disorder, protein biosynthesis: Transcription factor/coregulator deficiencies (1) Basic domains 1.2: Feingold syndrome · Saethre-Chotzen syndrome
1.3: Tietz syndrome(2) Zinc finger
DNA-binding domains2.1 (Intracellular receptor): Thyroid hormone resistance · Androgen insensitivity syndrome (PAIS, MAIS, CAIS) · Kennedy's disease · PHA1AD pseudohypoaldosteronism · Estrogen insensitivity syndrome · X-linked adrenal hypoplasia congenita · MODY 1 · Familial partial lipodystrophy 3 · SF1 XY gonadal dysgenesis
2.2: Barakat syndrome · Tricho–rhino–phalangeal syndrome
2.3: Greig cephalopolysyndactyly syndrome/Pallister-Hall syndrome · Denys–Drash syndrome · Duane-radial ray syndrome · MODY 7 · MRX 89 · Townes–Brocks syndrome · Acrocallosal syndrome · Myotonic dystrophy 2
2.5: Autoimmune polyendocrine syndrome type 1(3) Helix-turn-helix domains 3.1: ARX (Ohtahara syndrome, Lissencephaly X2) · HLXB9 (Currarino syndrome) · HOXD13 (SPD1 Synpolydactyly) · IPF1 (MODY 4) · LMX1B (Nail–patella syndrome) · MSX1 (Tooth and nail syndrome, OFC5) · PITX2 (Axenfeld syndrome 1) · POU4F3 (DFNA15) · POU3F4 (DFNX2) · ZEB1 (Posterior polymorphous corneal dystrophy 3, Fuchs' dystrophy 3) · ZEB2 (Mowat-Wilson syndrome)
3.2: PAX2 (Papillorenal syndrome) · PAX3 (Waardenburg syndrome 1&3) · PAX4 (MODY 9) · PAX6 (Gillespie syndrome, Coloboma of optic nerve) · PAX8 (Congenital hypothyroidism 2) · PAX9 (STHAG3)
3.3: FOXC1 (Axenfeld syndrome 3, Iridogoniodysgenesis, dominant type) · FOXC2 (Lymphedema–distichiasis syndrome) · FOXE1 (Bamforth–Lazarus syndrome) · FOXE3 (Anterior segment mesenchymal dysgenesis) · FOXF1 (ACD/MPV) · FOXI1 (Enlarged vestibular aqueduct) · FOXL2 (Premature ovarian failure 3) · FOXP3 (IPEX)
3.5: IRF6 (Van der Woude syndrome, Popliteal pterygium syndrome)(4) β-Scaffold factors
with minor groove contacts4.2: Hyperimmunoglobulin E syndrome
4.3: Holt-Oram syndrome · Li-Fraumeni syndrome · Ulnar–mammary syndrome
4.7: Campomelic dysplasia · MODY 3 · MODY 5 · SF1 (SRY XY gonadal dysgenesis, Premature ovarian failure 7) · SOX10 (Waardenburg syndrome 4c, Yemenite deaf-blind hypopigmentation syndrome)
4.11: Cleidocranial dysostosis(0) Other transcription factors 0.6: Kabuki syndromeUngrouped Transcription coregulators Categories:- Transcription factor deficiencies
- Intersexuality
- Syndromes
- Endocrine gonad disorders
- Androgen enters the cell.




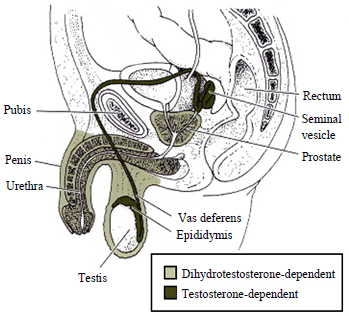

Wikimedia Foundation. 2010.