- Adrenoleukodystrophy
-
Adrenoleukodystrophy Classification and external resources 
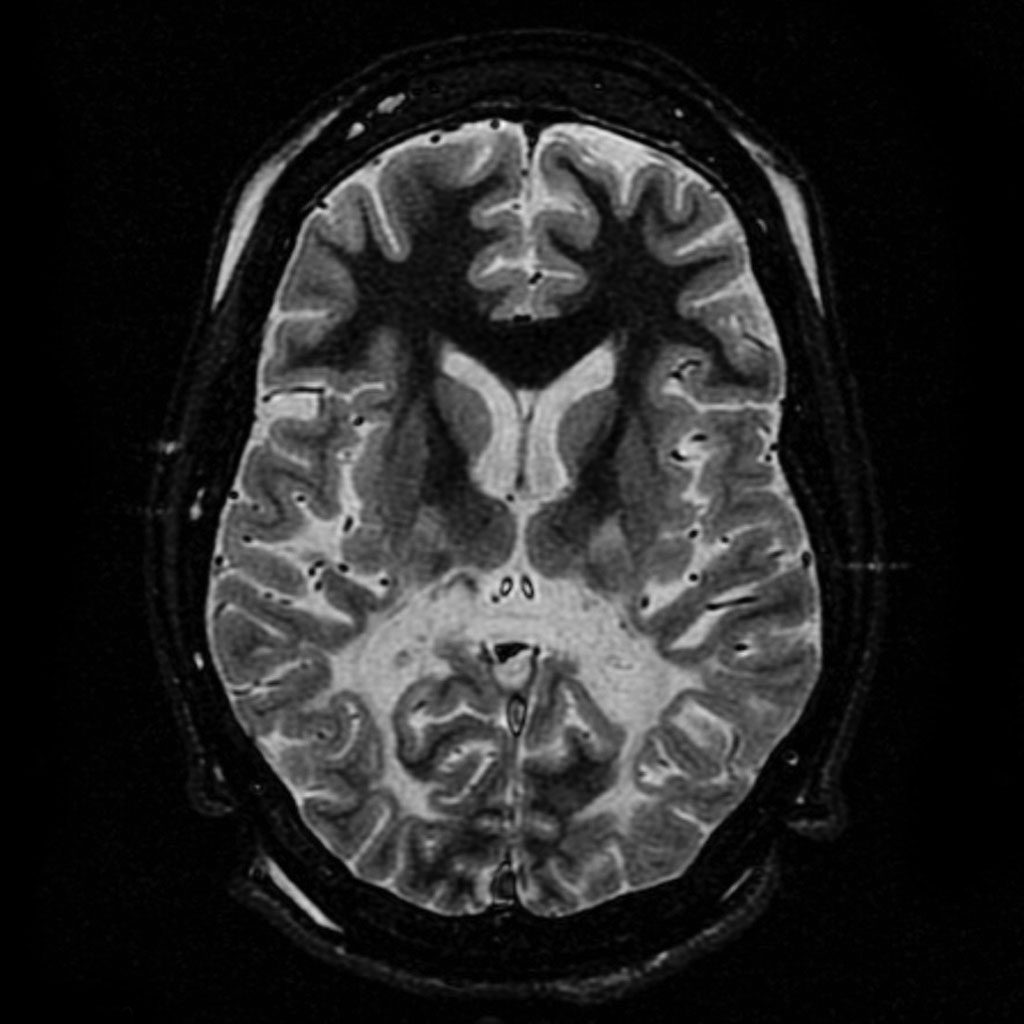
T2 weighted axial scan at the level of the caudate heads demonstrates marked loss of posterior white matter, with reduced volume and increased signal intensity. The anterior white matter is spared. Features are consistent with X-linked adrenoleukodystrophy.ICD-10 E71.3 ICD-9 330.0, 277.86 OMIM 300100 202370 DiseasesDB 292 MeSH D000326 GeneReviews X-Linked Adrenoleukodystrophy Adrenoleukodystrophy (ALD, also called Siemerling-Creutzfeldt Disease or Schilder's disease[1]:545) is a rare, inherited disorder that leads to progressive brain damage, failure of the adrenal glands and eventually death. ALD is a disease in a group of genetic disorders called leukodystrophies, whose chief feature is damage to myelin. In adrenoleukodystrophy, over-accumulation of VLCFAs leads to damage to the brain, adrenal gland, and peripheral nervous system.
on the age of onset of the disease. The classical, severe type is the childhood cerebral form which, as an X-linked disease, affects males. Symptoms normally start between the ages of 4 and 10 and include loss of previously acquired neurologic abilities, seizures, ataxia, Addison's disease, and degeneration of visual and auditory function. It has been seen that infants that have been positively diagnosed by the age of 1 year old have usually become very ill by the age of 10 to 12 years and die soon after. This severe form of the disease was first described by Ernst Siemerling and Hans Gerhard Creutzfeldt.[2] A similar form can also occur in adolescents and very rarely in adults. Addison's disease can be an initial symptom of ALD, and many pediatric endocrinologists will measure very long chain free fatty acids in newly diagnosed males with this condition, as a screening test for ALD.
In another form of ALD, one that primarily strikes young men, the spinal cord dysfunction is more prominent and is therefore called adrenomyeloneuropathy, or "AMN." The patients usually present with weakness and numbness of the limbs. Most victims of this form are also males, although some female carriers exhibit symptoms of AMN.[3]
Adult and neonatal forms of the disease also exist but they are extremely rare. (These tend to affect both males and females and be inherited in an autosomal recessive manner.) Some patients may present with sole findings of adrenal insufficiency.[citation needed]
Childhood cerebral type:
- Changes in muscle tone, especially muscle spasms and spasticity
- Crossed eyes
- Decreased understanding of verbal communication
- Deterioration of handwriting
- Difficulty at school
- Difficulty understanding spoken material
- Hearing loss
- Hyperactivity
- Worsening nervous system deterioration
Lack of brain-to-body coordination Loss of control and temper in outbreaks (random abrupt berserk behavior)
- Coma
- Decreased fine motor control
- Paralysis
- Seizures
- Swallowing difficulties
- Visual Impairment (Blindness)[4]
Contents
Diagnosis
The diagnosis is established by clinical findings and the detection of serum very long-chain free fatty acid levels.[5] MRI examination reveals white matter abnormalities, and neuro-imaging findings of this disease are somewhat reminiscent of the findings of multiple sclerosis. Genetic testing for the analysis of the defective gene is available in some centers.
Neonatal screening may become available in the future, which may permit early diagnosis and treatment.[6]
Genetics
X-linked

X-linked ALD (X-ALD) is the most common form of ALD. In X-ALD, the defective ABCD1 gene resides on the X chromosome (Xq28). The incidence of X-ALD is at least 1 in 20,000 male births.[7] The ABCD1 ("ATP-binding cassette, subfamily D, member 1") gene was discovered in 1993 and codes for a peroxisome membrane protein necessary for the β-oxidation of VLCFAs.[8]
X-ALD is characterized by excessive accumulation of very long-chain fatty acids (VLCFA), which are fatty acids with chains of 25–30 carbon atoms. The most common is hexacosanoate, with a 26 carbon skeleton. The elevation in (VLCFA) was originally described by Moser et al. in 1981.[9] The precise mechanisms through which high VLCFA concentrations in affected organs cause the disease is still unknown.
Autosomal
Neonatal adrenoleukodystrophy (NALD) is one of three autosomal recessive disorders which belong to the Zellweger spectrum of peroxisome biogenesis disorders (PBD-ZSD).[10] The other two disorders are Zellweger syndrome (ZS), and infantile Refsum disease (IRD).[11][12] NALD is most frequently caused by mutations in the PEX1, PEX5, PEX10, PEX13, and PEX26 genes.[13]
Treatment
While there is currently no cure for the disease, some dietary treatments, for example, a 4:1 mixture of glyceryl trioleate and glyceryl trierucate (Lorenzo's oil) in combination with a diet low in VLCSFA (very long chain saturated fatty acids), have been used with limited success, especially before disease symptoms appear. A 2005 study shows positive long-term results with this approach.[14] A 2007 report also appraises "Lorenzo's oil".[15] See also the Myelin Project. X-linked adrenoleukodystrophy has a very variable clinical course, even within a single family.[16] It is therefore not possible to determine if Lorenzo's oil is preventing progression of the disease in asymptomatic patients, or if these patients would have remained asymptomatic even without treatment. Current double blind placebo-controlled trials may be able to answer the questions regarding the effectiveness of treatment.
Hematopoietic stem cell transplantation (HSCT, including bone marrow transplant) is thought to be able to stop the progression of the X-ALD disease in asymptomatic or mildly symptomatic boys who have a Loes score lower than 9 (an MRI measure of the severity of the disease), though outcomes are markedly poorer in symptomatic boys.[17] HSCT carries a risk of mortality and morbidity and is not recommended for patients whose symptoms are already severe. Umbilical cord blood stem cell transplant may provide an alternative for patients who do not have a matched related stem cell donor. Preliminary studies suggest that the outcome of cord blood stem cell transplant for X-ALD is particularly good in very young, presymptomatic patients.[18]
Lovastatin is an anti-cholesterol drug that appears to have some effect in vitro, but not in mice with the animal model of adrenoleukodystrophy.[19] A clinical study of lovastatin showed encouraging biochemical changes, but no objective clinical improvement.[20] In a randomized, double-blind, placebo-controlled crossover trial, researchers found no effect of lovastatin on tissue levels of very long chain free fatty acids, and they recommended that it not be used in X-ALD.[21]
Currently, researchers at The Children's Hospital at the University of Minnesota, Dr. Charnas and Dr. Orchard, are investigating Mucomyst as an adjunct to bone marrow transplant, with some increase in survival time after transplant in 3 patients.[22]
According to a 1986 study, Oleic acid may lower the levels of saturated VLCFAs in vitro.[23]
Adrenal function must be tested periodically in all patients with ALD. Treatment with adrenal hormones can be lifesaving. Symptomatic and supportive treatments for ALD include physical therapy, psychological support, and special education. Recent evidence suggests that a mixture of oleic acid and erucic acid, known as "Lorenzo's Oil," administered to boys with X-ALD can reduce or delay the appearance of symptoms. Bone marrow transplants can provide long-term benefit to boys who have early evidence of X-ALD, but the procedure carries risk of mortality and morbidity and is not recommended for those whose symptoms are already severe or who have the adult-onset or neonatal forms. Oral administration of docosahexanoic (DHA) may help infants and children with neonatal ALD.[24]
Prevention
Although there is no guaranteed method for preventing X-ALD, health care providers can recommend genetic counseling for prospective parents with a family history of X-ALD. Female carriers can be diagnosed 85% of the time based on blood very long chain fatty acid (VLCFA) levels and genetic testing performed by specialized laboratories.
Prenatal diagnosis of X-ALD is also available. It is done by evaluating cells from chronic villus sampling or amniocentesis.[25]
Research
Active clinical trials are currently in progress to determine if the proposed treatments are effective:[26]
- Glyceryl Trioleate (Lorenzo's oil) for Adrenomyelneuropathy.[27]
- Beta Interferon and Thalidomide[28] This study is closed.
- Combination of Glyceryl Trierucate and Glyceryl Trioleate (Lorenzo's Oil) in asymptomatic patients.[29]
- Hematopoietic stem cell transplantation[30]
The National Institute of Neurological Disorders and Stroke (NINDS), supports the research on genetic disorders such as ALD. The goal of this research is to find ways to prevent, treat and cure these disorders. Intensive basic research has propsed two new approaches; 4-phenylbutyrate and lovastatin. These two new approaches could potentially lower levels of VLCFA in the brain, the therapeutic trials for both agents are planned.[31]
Experimental Therapies
In November 2009, Science published a report on a pilot study of two patients receiving gene therapy combined with blood-stem-cell therapy, and stating that the combination "may be a useful tool for treating" X-linked ALD.[32]
Presently, a boy with X-linked ALD (X-ALD) is undergoing gene therapy treatments in France. Professor Patrick Aubourg and Doctor Nathalie Cartier-Lacave are treating him with stem cells which have been altered by the introduction of replication-defective HIV-1–derived lentiviral vectors. They have also successfully treated other children in this way. Says Nathalie Cartier-Lacave, “We have completely stabilised the evolution of this disease. These children are well, they go to school, they have a social life, a normal family life, and there’s no reason to think that this stabilisation isn’t permanent.” The technique was developed in the Faculty of Pharmacy in Paris. First, peripheral blood mononuclear cells (PBMCs) were obtained from the X-ALD patients previously injected with granulocyte colony-stimulating factor. CD34+ cells (i.e. expressing the CD34 cell surface protein) were isolated from these PBMC cell population and then pre-activated ex vivo (outside the body) with a mixture of cytokines. Afterwards, a normal copy of the ABCD1 gene was introduced into these cells using lentiviral vectors. In parallel, patients were treated with cyclophosphamide and busulfan so that their hematopoietic stem cells (HSCs) containing the ABCD1 mutation were greatly diminished. The patients were then infused with their own CD34+ cells engineered to contain normal copies of the ABCD1 gene. These engineered CD34+ can differentiate into a vast number of different blood cell types. Most importantly, they turned into microglial cells that corrected the deficiencies in the brain responsible for X-ALD. If further research confirms these results, this could become standard treatment in the future – replacing heterologous bone marrow transplants which are risky, invasive, and which require a compatible donor to be effective.[33]
Prognosis
Treatment is symptomatic. Progressive neurological degeneration makes the prognosis generally poor. Death occurs within one to ten years of presentation of symptoms. The use of Lorenzo's Oil, bone marrow transplant, and gene therapy is currently under investigation.
Lorenzo Odone
Main article: Lorenzo OdoneLorenzo Michael Murphy Odone (May 29, 1978 – May 30, 2008) was probably the most famous patient with ALD. His parents Augusto and Michaela Odone, frustrated by the limited treatment available,[34] sparked the invention of "Lorenzo's oil", which is still being studied to see if it can prevent or delay the onset of disease. The quest for a treatment for Lorenzo was depicted in the 1992 film Lorenzo's Oil, and was the subject of the Phil Collins song "Lorenzo" (on his 1996 album Dance Into The Light).
References
- ^ James, William D.; Berger, Timothy G. (2006). Andrews' Diseases of the Skin: clinical Dermatology. Saunders Elsevier. ISBN 0-7216-2921-0.
- ^ Siemerling E, Creutzfeldt HG (1923). "Bronzekrankheit und sklerosierende Encephalomyelitis". Arch. Psychiat. Neurokrankh. 68: 217–44. doi:10.1007/BF01835678.
- ^ O'Brien TJ, Gates PG, Byrne E (April 1996). "Symptomatic female heterozygotes for adrenoleukodystrophy: A report of two unrelated cases and review of the literature". Journal of Clinical Neuroscience 3 (2): 166–70. doi:10.1016/S0967-5868(96)90012-0. PMID 18638861. http://linkinghub.elsevier.com/retrieve/pii/S0967-5868(96)90012-0.
- ^ "Adrenoleukodystrophy". The New York TImes. November 11, 2009.
- ^ Moser HW, Moser AB, Frayer KK et al. (October 1981). "Adrenoleukodystrophy: increased plasma content of saturated very long chain fatty acids". Neurology 31 (10): 1241–9. PMID 7202134.
- ^ Moser HW, Raymond GV, Dubey P (Dec 2005). "Adrenoleukodystrophy: new approaches to a neurodegenerative disease". JAMA 294 (24): 3131–4. doi:10.1001/jama.294.24.3131. PMID 16380594.
- ^ Berger, J.; Gartner, J. (2006). "X-linked adrenoleukodystrophy: Clinical, biochemical and pathogenetic aspects". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1763 (12): 1721. doi:10.1016/j.bbamcr.2006.07.010.
- ^ Mosser J, Douar AM, Sarde CO et al. (Feb 1993). "Putative X-linked adrenoleukodystrophy gene shares unexpected homology with ABC transporters". Nature 361 (6414): 726–30. doi:10.1038/361726a0. PMID 8441467.
- ^ Moser HW, Moser AB, Frayer KK et al. (Oct 1981). "Adrenoleukodystrophy: increased plasma content of saturated very long chain fatty acids". Neurology 31 (10): 1241–9. PMID 7202134.
- ^ Steinberg, S.; Dodt, G.; Raymond, G.; Braverman, N.; Moser, A.; Moser, H. (2006). "Peroxisome biogenesis disorders". Biochimica et Biophysica Acta (BBA) - Molecular Cell Research 1763 (12): 1733. doi:10.1016/j.bbamcr.2006.09.010.
- ^ GeneReviews: Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum
- ^ Krause, C.; Rosewich, H.; Thanos, M.; Gärtner, J. (2006). "Identification of novel mutations inPEX2,PEX6,PEX10,PEX12, andPEX13in Zellweger spectrum patients". Human Mutation 27 (11): 1157. doi:10.1002/humu.9462.
- ^ Online 'Mendelian Inheritance in Man' (OMIM) ADRENOLEUKODYSTROPHY, AUTOSOMAL NEONATAL FORM -202370
- ^ Moser, HW; Raymond GV, Lu S-E, Muenz LR, Moser AB, Xu J, Jones RO, Loes DJ, Melhem ER, Dubey P, Bezman L, Brereton NH, Odone A (2005-07). "Follow-up of 89 asymptomatic patients with adrenoleukodystrophy treated with Lorenzo's Oil". Archives of Neurology 62 (7): p. 1073–80. doi:10.1001/archneur.62.7.1073. PMID 16009761.
- ^ Moser HW, Moser AB, Hollandsworth K, Brereton NH, Raymond GV (Sep 2007). ""Lorenzo's oil" therapy for X-linked adrenoleukodystrophy: rationale and current assessment of efficacy". J. Mol. Neurosci. 33 (1): 105–13. doi:10.1007/s12031-007-0041-4. PMID 17901554.
- ^ Online 'Mendelian Inheritance in Man' (OMIM) Adrenoleukodystrophy -300100
- ^ Peters C, Charnas LR, Tan Y, Ziegler RS, Shapiro EG, DeFor T, Grewal SS, Orchard PJ, Abel SL, Goldman AI, Ramsay NK, Dusenbery KE, Loes DJ, Lockman LA, Kato S, Aubourg PR, Moser HW, Krivit W (2004). "Cerebral X-linked adrenoleukodystrophy: the international hematopoietic cell transplantation experience from 1982 to 1999". Blood 104 (3): 881–8. doi:10.1182/blood-2003-10-3402. PMID 15073029.
- ^ Beam D, Poe MD, Provenzale JM, Szabolcs P, Martin PL, Prasad V, Parikh S, Driscoll T, Mukundan S, Kurtzberg J, Escolar ML (2007). "Outcomes of unrelated umbilical cord blood transplantation for X-linked adrenoleukodystrophy". Biol Blood Marrow Transplant 13 (6): 665–74. doi:10.1016/j.bbmt.2007.01.082. PMID 17531776.
- ^ Yamada T, Shinnoh N, Taniwaki T et al. (September 2000). "Lovastatin does not correct the accumulation of very long-chain fatty acids in tissues of adrenoleukodystrophy protein-deficient mice". J. Inherit. Metab. Dis. 23 (6): 607–14. doi:10.1023/A:1005634130286. PMID 11032335. http://www.kluweronline.com/art.pdf?issn=0141-8955&volume=23&page=607.
- ^ Pai GS, Khan M, Barbosa E, Key LL, Craver JR, Curé JK, Betros R, Singh I (April 2000). "Lovastatin therapy for X-linked adrenoleukodystrophy: clinical and biochemical observations on 12 patients". Molecular Genetics and Metabolism 69 (4): 312–22. doi:10.1006/mgme.2000.2977. PMID 10870849. http://linkinghub.elsevier.com/retrieve/pii/S1096719200929779.
- ^ Engelen M, Ofman R, Dijkgraaf MG et al. (January 2010). "Lovastatin in X-linked adrenoleukodystrophy". The New England Journal of Medicine 362 (3): 276–7. doi:10.1056/NEJMc0907735. PMID 20089986.Lovaststin should not be prescribed as therapy to lower levels of very-long-chain fatty acids in patients with X-ALD.
- ^ Tolar J, Orchard PJ, Bjoraker KJ, Ziegler RS, Shapiro EG, Charnas L (Feb 2007). "N-acetyl-L-cysteine improves outcome of advanced cerebral adrenoleukodystrophy". Bone Marrow Transplant 39 (4): 211–5. doi:10.1038/sj.bmt.1705571. PMID 17290278.
- ^ Rizzo WB, Watkins PA, Phillips MW, Cranin D, Campbell B, Avigan J (March 1986). "Adrenoleukodystrophy: oleic acid lowers fibroblast saturated C22-26 fatty acids". Neurology 36 (3): 357–61. PMID 3951702.
- ^ "NINDS Adrenoleukodystrophy Information Page". National Institute of Neurological Disorders and Stroke. http://www.ninds.nih.gov/disorders/adrenoleukodystrophy/adrenoleukodystrophy.htm. Retrieved March 18, 2009.
- ^ Kaneshiro, Neil. "Adrenoleukodystrophy". http://www.ncbi.nlm.nih.gov/pubmedhealth/PMH0002165/. Retrieved 11/2/2009.
- ^ clinicaltrials.gov/
- ^ "A Phase III Trial of Lorenzo's Oil in Adrenomyeloneuropathy". http://www.clinicaltrials.gov/show/NCT00545597. Retrieved 2009-06-06.
- ^ ClinicalTrials.gov NCT00004450
- ^ "Study of Glyceryl Trierucate and glyceryl trioleate (Lorenzo's Oil) therapy in male children with adrenoleukodystrophy". http://www.clinicaltrials.gov/show/NCT00004418. Retrieved 2009-06-06.
- ^ "HSCT for High Risk Inherited Inborn Errors". http://www.clinicaltrials.gov/show/NCT00383448. Retrieved 2009-06-06.
- ^ "NINDS Adrenoleukodyestrophy Information Pag". http://www.ninds.nih.gov/disorders/adrenoleukodystrophy/adrenoleukodystrophy.htm. Retrieved March 18, 2009.
- ^ "Gene Therapy Technique Slows Brain Disease ALD Featured In Movie 'Lorenzo's Oil'" at Science News site
- ^ "Hematopoietic Stem Cell Gene Therapy with a Lentiviral Vector in X-Linked Adrenoleukodystrophy" at Science Mag site
- ^ "About Lorenzo, his Parents, and Oumouri". The Myelin Project. Archived from the original on 2006-04-27. http://web.archive.org/web/20060427045621/http://www.myelin.org/aboutlorenzo.htm. Retrieved 2006-06-03.
External links
- GeneReviews/NCBI/NIH/UW entry on Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum
- OMIM entries on Peroxisome Biogenesis Disorders, Zellweger Syndrome Spectrum
- European Leukodystrophy Foundation
- March of Dimes Foundation
- United Leukodystrophy Foundation
- Adrenoleukodystrophy at the Open Directory Project
- adrenoleukodystrophy at NINDS
- Images of ALD at USUHS
- Adrenoleukodystrophy at National Center for Biotechnology Information
- Information from ALDlife.org
Inborn error of lipid metabolism: fatty-acid metabolism disorders (E71.3, 277.81–277.85) Synthesis Degradation Acyl transportGeneralOtherTo acetyl-CoASjögren–Larsson syndromeGenetic disorder, organelle: Peroxisomal disorders and lysosomal structural disorders (E80.3, 277.86) Peroxisome biogenesis disorder Zellweger syndrome · Autosomal adrenoleukodystrophy · Infantile Refsum disease · Adult Refsum disease-2 · RCP 1Enzyme-related Transporter-related X-linked adrenoleukodystrophy/AdrenomyeloneuropathyLysosomal See also: proteins, intermediatesB structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkSex linkage: X-linked disorders X-linked recessive Immune Chronic granulomatous disease (CYBB) · Wiskott–Aldrich syndrome · X-linked severe combined immunodeficiency · X-linked agammaglobulinemia · Hyper-IgM syndrome type 1 · IPEX · X-linked lymphoproliferative disease · Properdin deficiencyHematologic Endocrine Metabolic amino acid: Ornithine transcarbamylase deficiency · Oculocerebrorenal syndrome
dyslipidemia: Adrenoleukodystrophy
carbohydrate metabolism: Glucose-6-phosphate dehydrogenase deficiency · Pyruvate dehydrogenase deficiency · Danon disease/glycogen storage disease Type IIb
lipid storage disorder: Fabry's disease
mucopolysaccharidosis: Hunter syndrome
purine-pyrimidine metabolism: Lesch–Nyhan syndrome
mineral: Menkes disease/Occipital horn syndromeNervous system X-Linked mental retardation: Coffin–Lowry syndrome · MASA syndrome · X-linked alpha thalassemia mental retardation syndrome · Siderius X-linked mental retardation syndrome
eye disorders: Color blindness (red and green, but not blue) · Ocular albinism (1) · Norrie disease · Choroideremia
other: Charcot–Marie–Tooth disease (CMTX2-3) · Pelizaeus–Merzbacher disease · SMAX2Skin and related tissue Dyskeratosis congenita · Hypohidrotic ectodermal dysplasia (EDA) ·
X-linked ichthyosis · X-linked endothelial corneal dystrophyNeuromuscular Urologic Bone/tooth No primary system Barth syndrome · McLeod syndrome · Smith-Fineman-Myers syndrome · Simpson–Golabi–Behmel syndrome · Mohr–Tranebjærg syndrome · Nasodigitoacoustic syndromeX-linked dominant X-linked hypophosphatemia · Focal dermal hypoplasia · Fragile X syndrome · Aicardi syndrome · Incontinentia pigmenti · Rett syndrome · CHILD syndrome · Lujan–Fryns syndrome · Orofaciodigital syndrome 1Genetic disorder, membrane: ABC-transporter disorders ABCA ABCA1 (Tangier disease) · ABCA3 (Surfactant metabolism dysfunction 3) · ABCA4 (Stargardt disease 1, Retinitis pigmentosa 19) · ABCA12 (Harlequin-type ichthyosis, Lamellar ichthyosis 2)ABCB ABCC ABCC2 (Dubin–Johnson syndrome) · ABCC6 (Pseudoxanthoma elasticum) · ABCC7 (Cystic fibrosis) · ABCC8 (HHF1, TNDM2) · ABCC9 (Dilated cardiomyopathy 1O)ABCD ABCD1 (Adrenoleukodystrophy, Adrenomyeloneuropathy)ABCG see also ABC transporters
B structural (perx, skel, cili, mito, nucl, sclr) · DNA/RNA/protein synthesis (drep, trfc, tscr, tltn) · membrane (icha, slcr, atpa, abct, othr) · transduction (iter, csrc, itra), trfkCategories:- Membrane transport protein disorders
- Leukodystrophies
- Demyelinating diseases of CNS
- Skin conditions resulting from errors in metabolism
- Neurological disorders in children
- Adrenal gland disorders
- Rare diseases
- Fatty-acid metabolism disorders
- Peroxisomal disorders

Wikimedia Foundation. 2010.