- 5-HT3 antagonist
-
5-HT3 receptor antagonists Drug class 
Skeletal formula of ondansetron, the prototypical 5-HT3 antagonistATC code A04AA MeSH D058831 AHFS/Drugs.com list of generics Consumer Reports best-buy-drugs WebMD Biological target 5-HT3 receptor The 5-HT3 antagonists are a class of medications that act as receptor antagonists at the 5-HT3 receptor, a subtype of serotonin receptor found in terminals of the vagus nerve and in certain areas of the brain. With the notable exception of alosetron and cilansetron, which are used in the treatment of irritable bowel syndrome, all 5-HT3 antagonists are antiemetics, used in the prevention and treatment of nausea and vomiting. They are particularly effective in controlling the nausea and vomiting produced by cancer chemotherapy and are considered the gold standard for this purpose.[1]
The 5-HT3 antagonists may be identified by the suffix –setron,[2] and are classified under code A04AA of the WHO's Anatomical Therapeutic Chemical Classification System.
Contents
History
The history of the 5-HT3 receptor antagonists began in 1957, when J.H. Gaddum and Zuleika P. Picarelli at the University of Edinburgh proposed the existence of two serotonin receptor subtypes, the M and D receptors (thus named because their function could be blocked by morphine and Dibenzyline respectively), in a landmark paper.[3] The 5-HT3 receptor was later found to correspond to the M receptor.[4] In the 1970s, John Fozard proved that metoclopramide and cocaine were weak antagonists at the 5-HT3 (5-HT-M) receptor. Fozard and Maurice Gittos later synthesized MDL 72222, the first potent and truly selective 5-HT3 receptor antagonist.[5][6] The antiemetic effects of metoclopramide were found to be partially because of its serotonin antagonism.[7]
While Fozard was investigating cocaine analogues, workers at Sandoz identified the potent, selective 5-HT3 receptor antagonist ICS 205-930 from which the first marketed selective 5-HT3 receptor antagonists ondansetron and granisetron were developed, and approved in 1991 and 1993 respectively.[5][8] Several compounds related to MDL 72222 were synthesized which eventually resulted in approval of tropisetron in 1994 and dolasetron in 1997.[8] A new and improved 5-HT3 receptor antagonist, named palonosetron, was approved in 2003.[8] The development of selective 5-HT3 receptor antagonists was a dramatic improvement in the treatment of nausea and vomiting.[7] Ondansetron, granisetron, dolasetron and palonosetron are currently approved in the United States, and form the cornerstone of therapy for the control of acute emesis with chemotherapy agents with moderate to high emetogenic potential.[9]
Development
5-HT3 receptor antagonists or serotonin antagonists were first introduced in the early 1990s, and they have become the most widely used antiemetic drugs in chemotherapy.[10] They have also been proven safe and effective for treatment of postoperative nausea and vomiting.[7] Serotonin (5-HT) is found widely distributed throughout the gut and the central nervous system. In the gut, 5-HT is found mostly in mucosal enterochromaffin cells. Enterochromaffin cells are sensory transducers that release 5-HT to activate intrinsic (via 5-HT1P and 5-HT4 receptors) and extrinsic (via 5-HT3 receptors) primary afferent nerves.[11] Chemotherapeutic drugs for malignant disorders that cause vomiting have been found to cause release of large amounts of serotonin from enterochromaffin cells in the gut, serotonin acts on 5-HT3 receptors in the gut and brain stem.[11]
Mechanism of action
The 5-HT3 receptors are present in several critical sites involved in emesis, including vagal afferents, the solitary tract nucleus (STN), and the area postrema itself. Serotonin is released by the enterochromaffin cells of the small intestine in response to chemotherapeutic agents and may stimulate vagal afferents (via 5-HT3 receptors) to initiate the vomiting reflex. The 5-HT3 receptor antagonists suppress vomiting and nausea by inhibiting serotonin binding to the 5-HT3 receptors. The highest concentration of 5-HT3 receptors in the central nervous system (CNS) are found in the STN and chemoreceptor trigger zone (CTZ), and 5-HT3 antagonists may also suppress vomiting and nausea by acting at these sites.[10]
When patients undergo chemotherapy, serotonin is released from enterochromaffin cells by the cytotoxicity, the selective 5-HT3 receptor antagonists prevent the ability of serotonin to activate and sensitize gastrointestinal vagal-nerve terminals to other emetogenic substances released.[12]
The 5-HT3 receptor
The 5-HT3 (5-HT3) receptor belongs to the Cys-loop superfamily of ligand-gated ion channels (LGICs) and therefore differs structurally and functionally from all other 5-HT (serotonin) receptors which are G protein-coupled receptors.[4][13][14] This ion channel is cation-selective and mediates neuronal depolarization and excitation within the central and peripheral nervous systems.[4] The rapidly activating, desensitizing, inward current is predominantly carried by sodium and potassium ions.[13] 5-HT3 receptors have a negligible permeability to anions.[4]
The 5-HT3 receptor consists of five subunits that may be the same (homopentameric 5-HT3A receptors) or different (heteropentameric receptors, usually consisting of 5-HT3A and 5-HT3B receptor subunits).[4][13][15]
 Figure 1. The subunits are assembled as a pentamer (right) and each subunit has four transmembrane domains (left).
Figure 1. The subunits are assembled as a pentamer (right) and each subunit has four transmembrane domains (left).
The subunits surround a central ion channel in a pseudo-symmetric manner (Fig.1). Each subunit comprises an extracellular N-terminal domain, four transmembrane domains (M1-M4) connected by intracellular (M1-M2 and M3-M4) and extracellular loops (M2-M3) and an extracellular C-terminus (Fig.1).[4] The extracellular domain is the site of action of agonists and competitive antagonists because of ligand binding and the transmembrane domain controls the movement of ions across the cell membrane.[13] The human subunits 5-HT3A and 5-HT3B have been isolated and as well as sharing 41% amino acid sequence identity the location of their genes are in close proximity on the long arm of chromosome 11. The 5-HT3C, 5-HT3D and 5-HT3E subunits have not been isolated.[4]
Genes that code for the subunits of the 5-HT3 receptor have been identified. HTR3A and HTR3B for the 5-HT3A and 5-HT3B subunits and in addition HTR3C, HTR3D and HTR3E genes encoding 5-HT3C, 5-HT3D and 5-HT3E subunits. The latter three tend to show peripherally restricted pattern of expression, with high levels in the gut. In human duodenum and stomach, for example, 5-HT3C and 5-HT3E mRNA might be greater than for 5-HT3A and 5-HT3B. There is some evidence to suggest that the 5-HT3 receptor subunits are an important contribution to the effectiveness of these compounds.[13] In patients treated with chemotherapeutic drugs, certain polymorphism of the HTR3B gene could predict successful antiemetic treatment. This could indicate that the 5-HT3B receptor subunit could be used as biomarker of antiemetic drug efficacy. HTR3C and HTR3E do not seem to form functional homomeric channels, but when co-expressed with HTR3A they form heteromeric complex with decreased or increased 5-HT efficacies. The pathophysiological role for these additional subunits has yet to be identified.[12]
Drug design
The 5-HT3 receptor ligand binding site
 Fig 2. Granisetron in the 5-HT3 receptor binding pocket with its aromatic rings between W183 and Y234 and its azabicyclic ring between W90 and F226
Fig 2. Granisetron in the 5-HT3 receptor binding pocket with its aromatic rings between W183 and Y234 and its azabicyclic ring between W90 and F226Experiments have shown evidence that the ligand-binding site is located at the interface of two adjacent subunits.[16] The ligand binding site is formed by three loops (A-C) from the principal ligand binding subunit (principal face) and three β-strands (D-F) from the adjacent subunit (complementary face).[4][14] The amino acid residue E129 on loop A faces into the binding pocket and forms a critical hydrogen bond with the hydroxyl group of 5-HT. Loop B contains W183, a critical tryptophan ligand binding residue that contributes to a cation-π interaction between the pi electron density of tryptophan and the primary amine of 5-HT. Loop C residues have been considered as candidates for the differing pharmacology of rodent and human 5-HT3 receptors because of their divergence between species. The most important aromatic residue within loop C is probably Y234 that lies opposite to the loop B tryptophan in the ligand binding pocket and is involved in ligand binding. Loops D and F are in fact β-strands not loops. W90 in loop D is critical for ligand binding and antagonists may directly contact R92. The azabicyclic ring of the competitive antagonist granisetron is located close to R92 and the aromatic rings lie close to W90. Loop E residues Y143, G148, E149, V150, Q151, N152, Y153 and K154 may be important for granisetron binding. The structure of loop F has yet to be clarified but W195 and D204 seem to be critical for ligand binding.[4] The results of several studies suggest that the orientation of granisetron (a 5-HT3 receptor antagonist )in the 5-HT3 receptor binding pocket is with its aromatic rings between Trp-183 and Tyr-234 and its azabicyclic ring between Trp-90 and Phe-226.(Fig.2)[13]
Binding affinity of 5-HT3 receptor antagonist[13] 5-HT3 receptor antagonists Binding affinity (Kd, Ki, K50) Species Tropisetron 11nM Human Granisetron 1,44 nM Human Ondansetron 4,9 nM Human Palonosetron 31,6 nM Rat cerebral cortex, Rabbit ileal myenteric plexus, Guinea-pig ileal plexus Dolasetron 20,03 nM NG 108-15 Metoclopramide (non-selective) 355 nM Human Cocaine 2,45-83 nM Rat-Rabbit The 5-HT3 receptor antagonists structure
 Fig 3. Ondansetron: First generation 5-HT3 receptor antagonist
Fig 3. Ondansetron: First generation 5-HT3 receptor antagonist Fig 4. Palonosetron: Second generation 5-HT3 receptor antagonist
Fig 4. Palonosetron: Second generation 5-HT3 receptor antagonistChemical structures of the first generation 5-HT3 receptor antagonist can be categorized to three main classes[7]
- Carbazole derivatives (ondansetron)
- Indazoles (Granisetron)
- Indoles (Tropisetron and Dolasetron)
The first-generation 5-HT3 receptor antagonist (ondansetron, dolasetron, granisetron, and tropisetron) have been the most important drugs in antiemetic therapy for emetogenic chemotherapy. They are especially effective in treating acute emesis, occurring in the first 24 hours following chemotherapy.[9] A newer drug palonosetron is a pharmacologically distinct and highly selective, second generation 5-HT3 receptor antagonist.[17] Palonosetron has two stereogenic centers and exists as four stereoisomers.[17] Palonosetron has longer half-life (40h) and greater receptor binding affinity (>30 fold; when compared to first generation antagonists).[9]
The 5-HT3 receptor antagonists pharmacophore
 Fig 5. The 5-HT3 receptor antagonists pharmacophore (schematic)
Fig 5. The 5-HT3 receptor antagonists pharmacophore (schematic)The pharmacophore of 5-HT3receptors consists of three components: a carbonyl-containing linking moiety, aromatic/heteroaromatic ring, and a basic center. The carbonyl group is coplanar to the aromatic ring. 5-HT3 receptor antagonists are more likely to bind in their protonated form.[18] Docking of a range of antagonists into a homology model of the 5-HT3 receptor binding site shows a reasonably good agreement with the pharmacophore model and supports the observed differences between species. Studies of granisetron in the binding pocket revealed that the aromatic rings of granisetron lie between W183 and Y234 and the azabiciclic ring between W90 and F226. In this study another energetically favorable location of granisetron was identified, closer to the membrane, on a position that could be a part of a binding/unbinding pathway for the ligand. A similarly located alternative binding site for granisetron has since been identified in another study of the 5-HT3 receptor.[13]
Structure-activity relationship (SAR)
 Fig. 6: The main pharmacophoric elements of the known 5-HT3 antagonist
Fig. 6: The main pharmacophoric elements of the known 5-HT3 antagonist5-HT3 receptor antagonists share the same pharmacophore.[13] An aromatic moiety (preferably indole), a linking acyl group capable of hydrogen bonding interactions, and a basic amine (nitrogen) can be regarded as the key pharmacophoric elements of the known 5-HT3receptor antagonists. There are steric limitations of the aromatic binding site and although two hydrogen-bonding interactions are possible on the heterocyclic linking group (oxadiazole capable of accepting two hydrogen bonds), only one is essential for high affinity. An optimal environment of the basic nitrogen is when its constrained within an azabicyclin system with the highes affinity observed for systems with nitrogen at the bridgehead position and secondary amines being more potent.[19] The 5-HT3 receptor can only accommodate small substituents on the charged amine, a methyl group being optimal.[13] The optimal distance between the aromatic binding site and the basic amine is 8,4-8,9 Å and it is best if a two-carbon linkage separates the oxadiazole and the nitrogen. An increasing substitution of R increases affinity.[19] The most potent antagonists of 5-HT3 receptors have a 6-membered aromatic ring, and they usually have 6,5 heterocyclic rings.[13] No correlation has been found between the lipophilicity of compounds and the 5-HT3 receptor affinities.[20] Since most of the known 5-HT3 antagonists are ester or amide derivatives they are potentially susceptible to hydrolysis, which could be avoided by incorporating H-bond acceptors within a 5-membered heteroaromatic ring.[19]
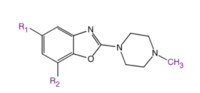 Fig. 7: The importance of C5 (R1) and C7(R2) substitution has been studied
Fig. 7: The importance of C5 (R1) and C7(R2) substitution has been studiedStructure-activity relationship (SAR) studies of LGIC receptor ligands are valuable to investigate their structure and function. An antagonist-like molecule with low intrinsic activity (ia) decreases the frequency of channel-opening and the permeability of ions. Small lipophilic C5 (R1) (see fig. 7) substituents afford compounds with potent antagonism which indicates that the C5 substituent may fit in a narrow, hydrophobic groove of the binding region in the receptor. It seems that the amino acid residues that interact with the C7 (R2) substituents have little to do with ligand binding but play a big role in ion channel gating. Sterically bulky substituents show a greater interaction with the gating amino acid residues and favor the open conformation af the ion channel because of sterical repulsion.[21]
 Fig. 8: The Carbonyl group is completely coplanar with the adjacent aromatic ring
Fig. 8: The Carbonyl group is completely coplanar with the adjacent aromatic ringOndansetron is a racemate but the stereochemistry of the asymmetric carbon atom is not an important factor in the 5-HT3 receptor interaction. Annelation of the 1,7-positions of the indole nucleus of ondansetron results in increased affinity for the receptor.[22]
A methyl- group appears to be as effective functionally as a chlorine in the R position (see fig. 8). The carbonyl group is responsible for a strong interaction with the receptor and contributes significantly to the binding process. This carbonyl group is completely coplanar with the adjacent aromatic ring, indicating that the receptor-bound conformation corresponds to one of the most stable conformations of this group in the flexible compounds.[18]
Comparative pharmacology of 5-HT3 antagonists
Despite that the 5-HT3 receptor antagonist share their mechanism of action, they have different chemical structures and exhibit differences in affinity for the receptor, dose response and duration of effect. Also they are metabolized in different ways, that is different components of the cytochrome P450 (CYP) system are predominate in the metabolism of the antagonists.[7]
The 5-HT3 receptor antagonist have similar activity. However patients who are resistant to one antagonist might benefit from another, possibly because the drugs are metabolized differently. A correlation exists between the number of active CYP 2D6 alleles and the number of vomiting episodes by patients who receive treatment with cisplatin and ondansetron or tropisetron. Patients with multiple alleles tend to be unresponsive to the antiemetic drug and vice versa.[12]
Comparative pharmacology of 5-HT3 receptor antagonist[10] Drug Chemical
natureReceptor antagonists T1/2 (h) Metabolism Dose Ondansetron Carbazole derivative 5-HT3 receptor antagonist and weak 5-HT4 antagonist 3.9 hours CYP1A1/2, CYP2D6, CYP 3A3/4/5 0.15 mg/kg Granisetron Indazole 5-HT3 receptor antagonist 9-11.6 hours CYP3A3/4/5 10 µg/kg Dolasetron Indole 5-HT3 receptor antagonist 7–9 hours CYP 3A3/4/5, CYP2D6 0.6–3 mg/kg Palonosetron Isoquinoline 5-HT3 receptor antagonist; highest affinity for 5-HT3 receptor in this class 40 hours CYP1A2, CYP2D6, CYP3A3/4/5[23] 0.25 mg x 1 dose Ramosetron Benzimidazole derivative 5-HT3 receptor antagonist 5.8 hours 300 µg/kg Tropisetron[7] Indole 5-HT3 receptor antagonist 5.6 hours CYP 3A3/4/5, CYP2D6 200 µg/kg Therapeutic uses
5-HT3 antagonists are most effective in the prevention and treatment of chemotherapy-induced nausea and vomiting (CINV), especially that caused by highly emetogenic drugs such as cisplatin; when used for this purpose, they may be given alone or, more frequently, with a glucocorticoid, usually dexamethasone. They are usually given intravenously, shortly before administration of the chemotherapeutic agent,[24] although some authors have argued that oral administration may be preferred.[25] The concomitant administration of a NK1 receptor antagonist, such as aprepitant, significantly increases the efficacy of 5-HT3 antagonists in preventing both acute and delayed CINV.[26]
The 5-HT3 antagonists are also indicated in the prevention and treatment of radiation-induced nausea and vomiting (RINV), when needed, and postoperative nausea and vomiting (PONV). Although they are more effective at controlling CINV—where they stop symptoms altogether in up to 70% of people, and reduce them in the remaining 30%—, they are just as effective as other agents for PONV.
Current evidence suggests that 5-HT3 antagonists are ineffective in controlling motion sickness.[27][28][29] A randomized, placebo-controlled trial of ondansetron to treat motion sickness in air ambulance personnel showed subjective improvement, but it was not statistically significant.[30]
Investigational
A small, open-label trial carried out in 2000 found ondansetron to be useful in treating antipsychotic-induced tardive dyskinesia in people with schizophrenia.[31][32] The study's patients also showed significant improvement in the disease's symptoms; a later double-blind, randomized controlled trial also found ondansetron to significantly improve schizophrenia symptoms when used as an adjunct to haloperidol, and people taking both drugs experienced fewer of the adverse effects commonly associated with haloperidol.[33]
Available agents
- Ondansetron (trade name Zofran in most countries) was the first 5-HT3 antagonist, developed by Glaxo around 1984. Its efficacy was first established in 1987, in animal models,[34][35] and it was extensively studied over the following years.[36] Ondansetron was approved by the U.S. Food and Drug Administration in 1991, and has since become available in several other countries, including the UK, Ireland, Australia, Canada, France and Brazil. As of 2008, ondansetron and granisetron are the only 5-HT3 antagonists available as a generic drug in the United States. Ondansetron may be given several times daily, depending on the severity of symptoms.
- Tropisetron (trade name Navoban) was also first described in 1984.[37] It is available in several countries, such as the UK, Australia and France, but not in the United States. The effects of tropisetron last up to 24 hours, so it only requires once-daily administration.
- Granisetron (trade name Kytril) was developed around 1988.[38] It is available in the U.S., UK, Australia and other countries. Clinical trials suggest that it is more effective than other 5-HT3 antagonists in preventing delayed CINV (nausea and vomiting that occur more than 24 hours after the first dose of chemotherapy).[39] It is taken once daily.
- Dolasetron (U.S. trade name Anzemet) was first mentioned in the literature in 1989.[40] It is a prodrug, and most of its effects are due to its active metabolite, hydrodolasetron, which is formed in the liver by the enzyme carbonyl reductase. Dolasetron was approved by the FDA in 1997, and is also administered once daily.
- Palonosetron (trade name Aloxi) is the newest 5-HT3 antagonist to become available in the U.S. market. It is an isoquinoline derivative, and is effective in preventing delayed CINV.[41] Palonosetron was approved by the FDA in 2003,[42] initially for intravenous use. An oral formulation was approved on August 22, 2008 for prevention of acute CINV alone, as a large clinical trial did not show oral administration to be as effective as IV use against delayed CINV.[43]
- Ramosetron (trade name Nasea) is only available in Japan and certain Southeast Asian countries as of 2008.[44] It has higher affinity for the 5-HT3 receptor than the older 5-HT3 antagonists, and maintains its effects over two days; it is therefore significantly more effective for delayed CINV.[45] In animal studies, ramosetron was also effective against irritable bowel syndrome-like symptoms.[46]
Alosetron and cilansetron—the latter being developed by Solvay—are not antiemetics; instead, they are indicated in the treatment of a subset of irritable bowel syndrome where diarrhea is the dominant symptom. Alosetron was withdrawn from the U.S. market in 2000 due to unacceptably frequent severe side effects, and is only available through a restrictive program to patients who meet certain requirements.[47]
Certain prokinetic drugs such as cisapride, renzapride and metoclopramide, although not 5-HT3 antagonists proper, possess some weak antagonist effect at the 5-HT3 receptor. Galanolactone, a diterpenoid found in ginger, is a 5-HT3 antagonist and is believed to at least partially mediate the anti-emetic activity of this plant.[48][49] Mirtazapine (trade name Remeron) is a tetracyclic antidepressant with 5-HT3 antagonist effects and strong anti-emetic properties. Studies show mirtazapine as equally effective in treating chemotherapy-related nausea and vomiting as standard treatments; it is also cheaper and has fewer side effects than typical anti-emetics, and its antidepressant qualities may be an added benefit for cancer populations.[50] Mirtazapine has also been used in the treatment of the motility disorder gastroparesis due to its anti-emetic effects.[51] Olanzapine (trade name Zyprexa), an atypical antipsychotic with anti-emetic properties similar to those of mirtazapine, also shows promise in treating chemotherapy-induced nausea and vomiting.[50]
Adverse effects
There are few side effects related to the use of 5-HT3 antagonists; the most common are constipation or diarrhea, headache, and dizziness.[52] Unlike antihistamines with antiemetic properties such as cyclizine, 5-HT3 antagonists do not produce sedation, nor do they cause extrapyramidal effects, as phenothiazines (such as prochlorperazine) sometimes do.
All 5-HT3 antagonists have been associated with asymptomatic electrocardiogram changes, such as prolongation of the PT and QTc intervals and certain arrhythmias.[52] The clinical significance of these side effects is unknown.
Pharmacokinetics
All 5-HT3 antagonists are well-absorbed and effective after oral administration,[25][52] and all are metabolized in the liver by various isoenzymes of the cytochrome P450 system. They do not, however, inhibit or induce these enzymes.[52]
Mechanism of action
As their name implies, 5-HT3 antagonists prevent serotonin from binding to 5-HT3 receptors. Such receptors are present mostly on the ends of afferent branches of the vagus nerve, which send signals directly to the brain's vomiting center in the medulla oblongata, and in the chemoreceptor trigger zone of the brain, which receives "input" from nausea-inducing agents in the bloodstream and communicates with the vomiting center. By preventing activation of these receptors, 5-HT3 antagonists interrupt one of the pathways that lead to vomiting.
The 5-HT3 antagonists are greatly selective, and have little affinity for other receptors, such as dopamine, histamine and muscarinic acetylcholine receptors.[52]
See also
Notes
- ^ de Wit R, Aapro M, Blower PR (2005). "Is there a pharmacological basis for differences in 5-HT3-receptor antagonist efficacy in refractory patients?". Cancer Chemother Pharmacol 56 (3): 231–8. doi:10.1007/s00280-005-1033-0. PMID 15838653.
- ^ World Health Organization (2006). The use of stems in the selection of International Nonproprietary Names (INN) for pharmaceutical substancesPDF (703 KiB). Geneva: WHO Press. Retrieved on 2007-05-15.
- ^ GADDUM JH, PICARELLI ZP (September 1957). "Two kinds of tryptamine receptor". British Journal of Pharmacology and Chemotherapy 12 (3): 323–8. PMC 1509685. PMID 13460238. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1509685.
- ^ a b c d e f g h i Barnes NM, Hales TG, Lummis SC, Peters JA (January 2009). "The 5-HT3 receptor--the relationship between structure and function". Neuropharmacology 56 (1): 273–84. doi:10.1016/j.neuropharm.2008.08.003. PMID 18761359.
- ^ a b King, Frank D.; Jones, Brian J.; Sanger, Gareth J. (1993). 5-Hydroxytryptamine-3 Receptor Antagonists. CRC Press. pp. 2–3. ISBN 978-0-8493-5463-2.
- ^ Galvan, M.; Gittos, M.; Fatmi, M. (October 1996). "DISCOVERY OF 5-HT3 RECEPTOR ANTAGONISTS AND DOLASETRON MESILATE". EJHP journal (6): 10–11. http://www.eahp.eu/EJHP/EJHP-Practice/Issue-6-1996/Science/DISCOVERY-OF-5-HT3-RECEPTOR-ANTAGONISTS-AND-DOLASETRON-MESILATE.
- ^ a b c d e f Gan TJ (2005). "Selective serotonin 5-HT3 receptor antagonists for postoperative nausea and vomiting: are they all the same?". CNS Drugs 19 (3): 225–38. doi:10.2165/00023210-200519030-00004. PMID 15740177.
- ^ a b c Billio, Atto; Clarke, Mike J.; Morello, Enrico; Billio, Atto (2006). Billio, Atto. ed. "Comparison of clinical efficacy of serotonin receptor antagonists in highly emetogenic chemotherapy". Cochrane Database of Systematic Reviews (4). doi:10.1002/14651858.CD006272.
- ^ a b c Oo TH, Hesketh PJ (April 2005). "Drug insight: New antiemetics in the management of chemotherapy-induced nausea and vomiting". Nature Clinical Practice. Oncology 2 (4): 196–201. doi:10.1038/ncponc0132. PMID 16264934.
- ^ a b c Brunton, Laurence L.; Lazo, John S.; Parker, Keith L. (2006). Goddman & Gilman's The Pharmacological Basis of Therapeutics. New York: McGraw-Hill. pp. 1000–1003. ISBN 0-07-1442280-3.
- ^ a b Kamm MA (March 2002). "Review article: the complexity of drug development for irritable bowel syndrome". Alimentary Pharmacology & Therapeutics 16 (3): 343–51. doi:10.1046/j.1365-2036.2002.01185.x. PMID 11876686.
- ^ a b c Sanger GJ (September 2008). "5-hydroxytryptamine and the gastrointestinal tract: where next?". Trends in Pharmacological Sciences 29 (9): 465–71. doi:10.1016/j.tips.2008.06.008. PMID 19086255.
- ^ a b c d e f g h i j k Thompson AJ, Lummis SC (2006). "5-HT3 Receptors". Current Pharmaceutical Design 12 (28): 3615–30. doi:10.2174/138161206778522029. PMC 2664614. PMID 17073663. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=2664614.
- ^ a b Reeves DC, Lummis SC (2002). "The molecular basis of the structure and function of the 5-HT3 receptor: a model ligand-gated ion channel (review)". Molecular Membrane Biology 19 (1): 11–26. doi:10.1080/09687680110110048. PMID 11989819.
- ^ Boess FG, Beroukhim R, Martin IL (March 1995). "Ultrastructure of the 5-hydroxytryptamine3 receptor". Journal of Neurochemistry 64 (3): 1401–5. doi:10.1046/j.1471-4159.1995.64031401.x. PMID 7861173.
- ^ Zhu LP, Ye DY, Tang Y (January 2007). "Structure-based 3D-QSAR studies on thiazoles as 5-HT3 receptor antagonists". Journal of Molecular Modeling 13 (1): 121–31. doi:10.1007/s00894-006-0131-1. PMID 16953442.
- ^ a b Tian K, Chen H, Tang J, Chen X, Hu Z (November 2006). "Enantioseparation of palonosetron hydrochloride by micellar electrokinetic chromatography with sodium cholate as chiral selector". Journal of Chromatography A 1132 (1–2): 333–6. doi:10.1016/j.chroma.2006.08.090. PMID 16999973.
- ^ a b Hibert MF, Hoffmann R, Miller RC, Carr AA (June 1990). "Conformation-activity relationship study of 5-HT3 receptor antagonists and a definition of a model for this receptor site". Journal of Medicinal Chemistry 33 (6): 1594–600. doi:10.1021/jm00168a011. PMID 2342053.
- ^ a b c Swain CJ, Baker R, Kneen C et al. (January 1991). "Novel 5-HT3 antagonists. Indole oxadiazoles". Journal of Medicinal Chemistry 34 (1): 140–51. doi:10.1021/jm00105a021. PMID 1992112.
- ^ Cappelli A, Donati A, Anzini M et al. (August 1996). "Molecular structure and dynamics of some potent 5-HT3 receptor antagonists. Insight into the interaction with the receptor". Bioorganic & Medicinal Chemistry 4 (8): 1255–69. doi:10.1016/0968-0896(96)00122-8. PMID 8879547.
- ^ Yoshida S, Watanabe T, Sato Y (May 2007). "Regulatory molecules for the 5-HT3 receptor ion channel gating system". Bioorganic & Medicinal Chemistry 15 (10): 3515–23. doi:10.1016/j.bmc.2007.02.054. PMID 17391967.
- ^ van Wijngaarden I, Hamminga D, van Hes R et al. (November 1993). "Development of high-affinity 5-HT3 receptor antagonists. Structure-affinity relationships of novel 1,7-annelated indole derivatives". Journal of Medicinal Chemistry 36 (23): 3693–9. doi:10.1021/jm00075a026. PMID 8246239.
- ^ Aapro M (2005). "5-HT(3)-receptor antagonists in the management of nausea and vomiting in cancer and cancer treatment". Oncology 69 (2): 97–109. doi:10.1159/000087979. PMID 16131816.
- ^ Herrstedt J, Aapro MS, Roila F, Kataja VV (2005). "ESMO Minimum Clinical Recommendations for prophylaxis of chemotherapy-induced nausea and vomiting (NV)".PDF Ann Oncol 16 Suppl 1: i77–9. PMID 15888767. doi:10.1093/annonc/mdi805
- ^ a b Lindley C, Blower P (2000). "Oral serotonin type 3-receptor antagonists for prevention of chemotherapy-induced emesis". Am J Health-Syst Pharm 57 (18): 1685–97. PMID 11006796. Free full text with registration at Medscape
- ^ Roila F, Fatigoni S (2006). "New antiemetic drugs" (PDF). Ann Oncol 17 Suppl 2: ii96–100. doi:10.1093/annonc/mdj936. PMID 16608997. http://annonc.oxfordjournals.org/cgi/reprint/17/suppl_2/ii96.
- ^ Stott JR, Barnes GR, Wright RJ, Ruddock CJ (1989). "The effect on motion sickness and oculomotor function of GR 38032F, a 5-HT3-receptor antagonist with anti-emetic properties". British journal of clinical pharmacology 27 (2): 147–57. PMC 1379774. PMID 2523720. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1379774.
- ^ Levine ME, Chillas JC, Stern RM, Knox GW (2000). "The effects of serotonin (5-HT3) receptor antagonists on gastric tachyarrhythmia and the symptoms of motion sickness". Aviat Space Environ Med 71 (11): 1111–4. PMID 11086664.
- ^ Muth ER, Elkins AN (July 2007). "High dose ondansetron for reducing motion sickness in highly susceptible subjects". Aviat Space Environ Med 78 (7): 686–92. PMID 17679566.
- ^ Dula D, Rosenbach S (2006). "A randomized clinical trial comparing ondansetron with placebo in aeromedical personel with motion sickness". Paper presented at the annual meeting of the National Association of EMS Physicians, Registry Resort, Naples, FL, January 19–21, 2006. Retrieved on April 25, 2009.
- ^ Zullino DF, Eap CB, Voirol P (2001). "Ondansetron for tardive dyskinesia". Am J Psychiatry 158 (4): 657–8. doi:10.1176/appi.ajp.158.4.657-a. PMID 11282718.
- ^ Sirota P, Mosheva T, Shabtay H, Giladi N, Korczyn AD (2000). "Use of the selective serotonin 3 receptor antagonist ondansetron in the treatment of neuroleptic-induced tardive dyskinesia". Am J Psychiatry 157 (2): 287–9. doi:10.1176/appi.ajp.157.2.287. PMID 10671405. Free full text
- ^ Zhang ZJ, Kang WH, Li Q, Wang XY, Yao SM, Ma AQ (2006). "Beneficial effects of ondansetron as an adjunct to haloperidol for chronic, treatment-resistant schizophrenia: a double-blind, randomized, placebo-controlled study". Schizophrenia Research 88 (1–3): 102–10. doi:10.1016/j.schres.2006.07.010. PMID 16959472.
- ^ Hagan RM, Butler A, Hill JM, Jordan CC, Ireland SJ, Tyers MB (1987). "Effect of the 5-HT3 receptor antagonist, GR38032F, on responses to injection of a neurokinin agonist into the ventral tegmental area of the rat brain". Eur. J. Pharmacol. 138 (2): 303–5. doi:10.1016/0014-2999(87)90450-X. PMID 2442006.
- ^ Costall B, Gunning SJ, Naylor RJ, Tyers MB (1987). "The effect of GR38032F, novel 5-HT3-receptor antagonist on gastric emptying in the guinea-pig". Br. J. Pharmacol. 91 (2): 263–4. PMC 1853517. PMID 2955843. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1853517.
- ^ See Eur J Cancer Clin Oncol 1989; 25 Suppl 1.
- ^ Donatsch P, Engel G, Richardson BP, Stadler PA (1984). "A highly selective and potent antagonist at peripheral neuronal 5-hydroxy tryptamine receptors". Br J Pharmacol 81: 34P.
- ^ Zussman BD, Clarkeson A, Coates PE, Rapeport WG (1988). "The pharmacokinetic profile of BRL 43694, a novel 5-HT3 receptor antagonist, in healthy male volunteers". Br J Clin Pharmacol 25: 107P.
- ^ Aapro M (2004). "Granisetron: an update on its clinical use in the management of nausea and vomiting". Oncologist 9 (6): 673–86. doi:10.1634/theoncologist.9-6-673. PMID 15561811. Free full text
- ^ Sorensen SM, Humphreys TM, Palfreyman MG (1989). "Effect of acute and chronic MDL 73,147EF, a 5-HT3 receptor antagonist, on A9 and A10 dopamine neurons". Eur. J. Pharmacol. 163 (1): 115–8. doi:10.1016/0014-2999(89)90402-0. PMID 2744086.
- ^ De Leon A (2006). "Palonosetron (Aloxi): a second-generation 5-HT3 receptor antagonist for chemotherapy-induced nausea and vomiting". Proceedings (Baylor University. Medical Center) 19 (4): 413–6. PMC 1618755. PMID 17106506. http://www.pubmedcentral.nih.gov/articlerender.fcgi?tool=pmcentrez&artid=1618755. Full text at PMC: 1618755.
- ^ "FDA Approves Aloxi (Palonosetron) For Treatment of Chemotherapy-Related Nausea and Vomiting" (Press release). Doctor's Guide Publishing Limited. July 28, 2003. http://www.docguide.com/news/content.nsf/news/8525697700573E1885256D71004DE600. Retrieved 2007-05-15.
- ^ Waknine, Yael (September 4, 2008). "FDA Approvals: Nplate, Aloxi, Vidaza". Medscape. http://www.medscape.com/viewarticle/580032. Retrieved 2008-09-04. Freely available with registration.
- ^ Abridged prescribing information - Nasea (MIMS Philippines). Retrieved on June 13, 2008.
- ^ Rabasseda X (February 2002). "Ramosetron, a 5-HT3 receptor antagonist for the control of nausea and vomiting". Drugs Today (Barc) 38 (2): 75–89. doi:10.1358/dot.2002.38.2.820104. PMID 12532186.
- ^ Hirata T, Funatsu T, Keto Y, Nakata M, Sasamata M (February 2007). "Pharmacological profile of ramosetron, a novel therapeutic agent for IBS". Inflammopharmacology 15 (1): 5–9. doi:10.1007/s10787-006-1537-1. PMID 17323187.
- ^ GlaxoSmithKline (2005). Lotronex Prescribing InformationPDF (203 KiB). U.S. Food and Drug Administration. Retrieved on 2009-07-30.
- ^ Ku, Valerie (2003). Ginger. University of Colorado at Denver and Health Sciences Center School of Pharmacy. Retrieved on 2007-10-25.
- ^ Huang QR, Iwamoto M, Aoki S et al. (1991). "Anti-5-hydroxytryptamine3 effect of galanolactone, diterpenoid isolated from ginger". Chem Pharm Bull 39 (2): 397–9. PMID 2054863.
- ^ a b Kast R E; Foley, KF (2007). "Cancer chemotherapy and cachexia: mirtazapine and olanzapine are 5-HT3 antagonists with good antinausea effects". European Journal of Cancer Care 16 (4): 351–354. doi:10.1111/j.1365-2354.2006.00760.x. PMID 17587360. http://www3.interscience.wiley.com/cgi-bin/fulltext/117989553/PDFSTART.
- ^ Kim S; Shin, IS; Kim, JM; Kang, HC; Mun, JU; Yang, SJ; Yoon, JS (2006). "Mirtazapine for Severe Gastroparesis Unresponsive to Conventional Prokinetic Treatment". Psychosomatics 47 (5): 440–442. doi:10.1176/appi.psy.47.5.440. PMID 16959934. http://psy.psychiatryonline.org/cgi/content/full/47/5/440.
- ^ a b c d e "5-Hydroxytryptamine3 (5-HT3) Receptor Antagonists". Oregon State University College of Pharmacy. 2003. http://pharmacy.oregonstate.edu/drug_policy/pages/dur_board/reviews/articles/hydroxytryptamine3.html. Retrieved 2007-05-15.
References
- Pasricha, Pankaj J. (2006). "Treatment of Disorders of Bowel Motility and Water Flux; Antiemetics; Agents Used in Biliary and Pancreatic Disease". In Laurence Brunton, John Lazo, Keith Parker (eds.). Goodman & Gilman's The Pharmacological Basis of Therapeutics (11th ed.). New York: McGraw-Hill. ISBN 978-0071422802.
- Hillier, Keith; Robert J. Naylor (2006). "Drugs and the Gastrointestinal System". In Clive Page, Brian Hoffmann, Michael Curtis, Michael Walker. Integrated Pharmacology (3rd ed.). Mosby. ISBN 978-0323040808.
Serotonergics 5-HT1 receptor ligands Agonists: Azapirones: Alnespirone • Binospirone • Buspirone • Enilospirone • Eptapirone • Gepirone • Ipsapirone • Perospirone • Revospirone • Tandospirone • Tiospirone • Umespirone • Zalospirone; Antidepressants: Etoperidone • Nefazodone • Trazodone • Vortioxetine; Antipsychotics: Aripiprazole • Asenapine • Clozapine • Quetiapine • Ziprasidone; Ergolines: Dihydroergotamine • Ergotamine • Lisuride • Methysergide • LSD; Tryptamines: 5-CT • 5-MeO-DMT • 5-MT • Bufotenin • DMT • Indorenate • Psilocin • Psilocybin; Others: 8-OH-DPAT • Adatanserin • Befiradol • BMY-14802 • Cannabidiol • Dimemebfe • Ebalzotan • Eltoprazine • F-11,461 • F-12,826 • F-13,714 • F-14,679 • F-15,063 • F-15,599 • Flesinoxan • Flibanserin • Lesopitron • LY-293,284 • LY-301,317 • MKC-242 • NBUMP • Osemozotan • Oxaflozane • Pardoprunox • Piclozotan • Rauwolscine • Repinotan • Roxindole • RU-24,969 • S 14,506 • S-14,671 • S-15,535 • Sarizotan • SSR-181,507 • Sunepitron • U-92,016-A • Urapidil • Vilazodone • Xaliproden • Yohimbine
Antagonists: Antipsychotics: Iloperidone • Risperidone • Sertindole; Beta blockers: Alprenolol • Cyanopindolol • Iodocyanopindolol • Oxprenolol • Pindobind • Pindolol • Propranolol • Tertatolol; Others: AV965 • BMY-7,378 • CSP-2503 • Dotarizine • Flopropione • GR-46611 • Isamoltane • Lecozotan • Mefway • Metitepine/Methiothepin • MPPF • NAN-190 • PRX-00023 • Robalzotan • S-15535 • SB-649,915 • SDZ 216-525 • Spiperone • Spiramide • Spiroxatrine • UH-301 • WAY-100,135 • WAY-100,635 • XylamidineAgonists: Lysergamides: Dihydroergotamine • Ergotamine • Methysergide; Piperazines: Eltoprazine • TFMPP; Triptans: Avitriptan • Eletriptan • Sumatriptan • Zolmitriptan; Tryptamines: 5-CT • 5-MT; Others: CGS-12066A • CP-93,129 • CP-94,253 • CP-135,807 • RU-24,969 • Vortioxetine
Antagonists: Lysergamides: Metergoline; Others: AR-A000002 • Elzasonan • GR-127,935 • Isamoltane • Metitepine/Methiothepin • SB-216,641 • SB-224,289 • SB-236,057 • YohimbineAgonists: Lysergamides: Dihydroergotamine • Methysergide; Triptans: Almotriptan • Avitriptan • Eletriptan • Frovatriptan • Naratriptan • Rizatriptan • Sumatriptan • Zolmitriptan; Tryptamines: 5-CT • 5-Ethyl-DMT • 5-MT • 5-(Nonyloxy)tryptamine; Others: CP-135,807 • CP-286,601 • GR-46611 • L-694,247 • L-772,405 • PNU-109,291 • PNU-142,633
Antagonists: Lysergamides: Metergoline; Others: Alniditan • BRL-15,572 • Elzasonan • GR-127,935 • Ketanserin • LY-310,762 • LY-367,642 • LY-456,219 • LY-456,220 • Metitepine/Methiothepin • Ritanserin • Yohimbine • ZiprasidoneAgonists: Lysergamides: Methysergide; Triptans: Eletriptan; Tryptamines: BRL-54443 • Tryptamine
Antagonists: Metitepine/MethiothepinAgonists: Triptans: Eletriptan • Naratriptan • Sumatriptan; Tryptamines: 5-MT; Others: BRL-54443 • Lasmiditan • LY-334,370
Antagonists: Metitepine/Methiothepin5-HT2 receptor ligands Agonists: Lysergamides: ALD-52 • Ergometrine • Lisuride • LA-SS-Az • LSD • LSD-Pip • Lysergic acid 2-butyl amide • Lysergic acid 3-pentyl amide • Methysergide; Phenethylamines: 25I-NBF • 25I-NBMD • 25I-NBOH • 25I-NBOMe • 2C-B • 2C-B-FLY • 2CB-Ind • 2C-C-NBOMe • 2C-E • 2C-I • 2C-TFM-NBOMe • 2C-T-2 • 2C-T-7 • 2C-T-21 • 2CBCB-NBOMe • 2CBFly-NBOMe • Bromo-DragonFLY • DOB • DOC • DOI • DOM • MDA • MDMA • Mescaline • TCB-2 • TFMFly; Piperazines: BZP • Quipazine • TFMPP; Tryptamines: 5-CT • 5-MeO-α-ET • 5-MeO-α-MT • 5-MeO-DET • 5-MeO-DiPT • 5-MeO-DMT • 5-MeO-DPT • 5-MT • α-ET • α-Methyl-5-HT • α-MT • Bufotenin • DET • DiPT • DMT • DPT • Psilocin • Psilocybin; Others: AL-34662 • AL-37350A • Dimemebfe • Medifoxamine • Oxaflozane • PNU-22394 • RH-34
Antagonists: Atypical antipsychotics: Amperozide • Aripiprazole • Carpipramine • Clocapramine • Clozapine • Gevotroline • Iloperidone • Melperone • Mosapramine • Olanzapine • Paliperidone • Pimozide • Quetiapine • Risperidone • Sertindole • Ziprasidone • Zotepine; Typical antipsychotics: Loxapine • Pipamperone; Antidepressants: Amitriptyline • Amoxapine • Aptazapine • Etoperidone • Mianserin • Mirtazapine • Nefazodone • Teniloxazine • Trazodone; Others: 5-I-R91150 • AC-90179 • Adatanserin • Altanserin • AMDA • APD-215 • Blonanserin • Cinanserin • CSP-2503 • Cyproheptadine • Deramciclane • Dotarizine • Eplivanserin • Esmirtazapine • Fananserin • Flibanserin • Ketanserin • KML-010 • Lubazodone • Mepiprazole • Metitepine/Methiothepin • Nantenine • Pimavanserin • Pizotifen • Pruvanserin • Rauwolscine • Ritanserin • S-14,671 • Sarpogrelate • Setoperone • Spiperone • Spiramide • SR-46349B • Volinanserin • Xylamidine • YohimbineAgonists: Oxazolines: 4-Methylaminorex • Aminorex; Phenethylamines: Chlorphentermine • Cloforex • DOB • DOC • DOI • DOM • Fenfluramine • MDA • MDMA • Norfenfluramine; Tryptamines: 5-CT • 5-MT • α-Methyl-5-HT; Others: BW-723C86 • Cabergoline • mCPP • Pergolide • PNU-22394 • Ro60-0175
Antagonists: Agomelatine • Asenapine • EGIS-7625 • Ketanserin • Lisuride • LY-272,015 • Metitepine/Methiothepin • PRX-08066 • Rauwolscine • Ritanserin • RS-127,445 • Sarpogrelate • SB-200,646 • SB-204,741 • SB-206,553 • SB-215,505 • SB-221,284 • SB-228,357 • SDZ SER-082 • Tegaserod • YohimbineAgonists: Phenethylamines: 2C-B • 2C-E • 2C-I • 2C-T-2 • 2C-T-7 • 2C-T-21 • DOB • DOC • DOI • DOM • MDA • MDMA • Mescaline; Piperazines: Aripiprazole • mCPP • TFMPP; Tryptamines: 5-CT • 5-MeO-α-ET • 5-MeO-α-MT • 5-MeO-DET • 5-MeO-DiPT • 5-MeO-DMT • 5-MeO-DPT • 5-MT • α-ET • α-Methyl-5-HT • α-MT • Bufotenin • DET • DiPT • DMT • DPT • Psilocin • Psilocybin; Others: A-372,159 • AL-38022A • CP-809,101 • Dimemebfe • Lorcaserin• Medifoxamine • MK-212 • Org 12,962 • ORG-37,684 • Oxaflozane • PNU-22394 • Ro60-0175 • Ro60-0213 • Vabicaserin • WAY-629 • WAY-161,503 • YM-348
Antagonists: Atypical antipsychotics: Clozapine • Iloperidone • Melperone • Olanzapine • Paliperidone • Pimozide • Quetiapine • Risperidone • Sertindole • Ziprasidone • Zotepine; Typical antipsychotics: Chlorpromazine • Loxapine • Pipamperone; Antidepressants: Agomelatine • Amitriptyline • Amoxapine • Aptazapine • Etoperidone • Fluoxetine • Mianserin • Mirtazapine • Nefazodone • Nortriptyline • Tedatioxetine • Trazodone; Others: Adatanserin • Cinanserin • Cyproheptadine • Deramciclane • Dotarizine • Eltoprazine • Esmirtazapine • FR-260,010 • Ketanserin • Ketotifen • Latrepirdine • Metitepine/Methiothepin • Methysergide • Pizotifen • Ritanserin • RS-102,221 • S-14,671 • SB-200,646 • SB-206,553 • SB-221,284 • SB-228,357 • SB-242,084 • SB-243,213 • SDZ SER-082 • Xylamidine5-HT3, 5-HT4, 5-HT5, 5-HT6, 5-HT7 ligands Agonists: Piperazines: BZP • Quipazine; Tryptamines: 2-Methyl-5-HT • 5-CT; Others: Chlorophenylbiguanide • Butanol • Ethanol • Halothane • Isoflurane • RS-56812 • SR-57,227 • SR-57,227-A • Toluene • Trichloroethane • Trichloroethanol • Trichloroethylene • YM-31636
Antagonists: Antiemetics: AS-8112 • Alosetron • Azasetron • Batanopride • Bemesetron • Cilansetron • Dazopride • Dolasetron • Granisetron • Lerisetron • Ondansetron • Palonosetron • Ramosetron • Renzapride • Tropisetron • Zacopride • Zatosetron; Atypical antipsychotics: Clozapine • Olanzapine • Quetiapine; Tetracyclic antidepressants: Amoxapine • Mianserin • Mirtazapine; Others: CSP-2503 • ICS-205,930 • MDL-72,222 • Memantine • Nitrous Oxide • Ricasetron • Sevoflurane • Tedatioxetine • Thujone • Vortioxetine • XenonAgonists: Gastroprokinetic Agents: Cinitapride • Cisapride • Dazopride • Metoclopramide • Mosapride • Prucalopride • Renzapride • Tegaserod • Velusetrag • Zacopride; Others: 5-MT • BIMU8 • CJ-033,466 • PRX-03140 • RS-67333 • RS-67506 • SL65.0155 • Antagonists: GR-113,808 • GR-125,487 • L-Lysine • Piboserod • RS-39604 • RS-67532 • SB-203,186 • SB-204,070Agonists: Lysergamides: Ergotamine • LSD; Tryptamines: 5-CT; Others: Valerenic Acid
Antagonists: Asenapine • Latrepirdine • Metitepine/Methiothepin • Ritanserin • SB-699,551
* Note that the 5-HT5B receptor is not functional in humans.Agonists: Lysergamides: Dihydroergotamine • Ergotamine • Lisuride • LSD • Mesulergine • Metergoline • Methysergide; Tryptamines: 2-Methyl-5-HT • 5-BT • 5-CT • 5-MT • Bufotenin • E-6801 • E-6837 • EMD-386,088 • EMDT • LY-586,713 • Tryptamine; Others: WAY-181,187 • WAY-208,466
Antagonists: Antidepressants: Amitriptyline • Amoxapine • Clomipramine • Doxepin • Mianserin • Nortriptyline; Atypical antipsychotics: Aripiprazole • Asenapine • Clozapine • Fluperlapine • Iloperidone • Olanzapine • Tiospirone; Typical antipsychotics: Chlorpromazine • Loxapine; Others: BGC20-760 • BVT-5182 • BVT-74316 • Cerlapirdine • EGIS-12,233 • GW-742,457 • Ketanserin • Latrepirdine • Lu AE58054 • Metitepine/Methiothepin • MS-245 • PRX-07034 • Ritanserin • Ro04-6790 • Ro 63-0563 • SB-258,585 • SB-271,046 • SB-357,134 • SB-399,885 • SB-742,457Agonists: Lysergamides: LSD; Tryptamines: 5-CT • 5-MT • Bufotenin; Others: 8-OH-DPAT • AS-19 • Bifeprunox • E-55888 • LP-12 • LP-44 • RU-24,969 • Sarizotan
Antagonists: Lysergamides: 2-Bromo-LSD • Bromocriptine • Dihydroergotamine • Ergotamine • Mesulergine • Metergoline • Methysergide; Antidepressants: Amitriptyline • Amoxapine • Clomipramine • Imipramine • Maprotiline • Mianserin; Atypical antipsychotics: Amisulpride • Aripiprazole • Clozapine • Olanzapine • Risperidone • Sertindole • Tiospirone • Ziprasidone • Zotepine; Typical antipsychotics: Chlorpromazine • Loxapine; Others: Butaclamol • EGIS-12,233 • Ketanserin • LY-215,840 • Metitepine/Methiothepin • Pimozide • Ritanserin • SB-258,719 • SB-258,741 • SB-269,970 • SB-656,104 • SB-656,104-A • SB-691,673 • SLV-313 • SLV-314 • Spiperone • SSR-181,507 • VortioxetineReuptake inhibitors Selective serotonin reuptake inhibitors (SSRIs): Alaproclate • Citalopram • Dapoxetine • Desmethylcitalopram • Desmethylsertraline • Escitalopram • Femoxetine • Fluoxetine • Fluvoxamine • Indalpine • Ifoxetine • Litoxetine • Lubazodone • Panuramine • Paroxetine • Pirandamine • RTI-353 • Seproxetine • Sertraline • Tedatioxetine • Vilazodone • Vortioxetine • Zimelidine; Serotonin-norepinephrine reuptake inhibitors (SNRIs): Bicifadine • Desvenlafaxine • Duloxetine • Eclanamine • Levomilnacipran • Milnacipran • Sibutramine • Venlafaxine; Serotonin-norepinephrine-dopamine reuptake inhibitors (SNDRIs): Brasofensine • Diclofensine • DOV-102,677 • DOV-21,947 • DOV-216,303 • NS-2359 • SEP-225289 • SEP-227,162 • Tesofensine; Tricyclic antidepressants (TCAs): Amitriptyline • Butriptyline • Cianopramine • Clomipramine • Desipramine • Dosulepin • Doxepin • Imipramine • Lofepramine • Nortriptyline • Pipofezine • Protriptyline • Trimipramine; Tetracyclic antidepressants (TeCAs): Amoxapine; Piperazines: Nefazodone • Trazodone; Antihistamines: Brompheniramine • Chlorphenamine • Diphenhydramine • Mepyramine/Pyrilamine • Pheniramine • Tripelennamine; Opioids: Pethidine • Methadone • Propoxyphene; Others: Cocaine • CP-39,332 • Cyclobenzaprine • Dextromethorphan • Dextrorphan • EXP-561 • Fezolamine • Mesembrine • Nefopam • PIM-35 • Pridefine • Roxindole • SB-649,915 • ZiprasidoneReleasing agents Aminoindanes: 5-IAI • AMMI • ETAI • MDAI • MDMAI • MMAI • TAI; Aminotetralins: 6-CAT • 8-OH-DPAT • MDAT • MDMAT; Oxazolines: 4-Methylaminorex • Aminorex • Clominorex • Fluminorex; Phenethylamines (also Amphetamines, Cathinones, Phentermines, etc): 2-Methyl-MDA • 4-CAB • 4-FA • 4-FMA • 4-HA • 4-MTA • 5-APDB • 5-Methyl-MDA • 6-APDB • 6-Methyl-MDA • AEMMA • Amiflamine • BDB • BOH • Brephedrone • Butylone • Chlorphentermine • Cloforex • Amfepramone • Metamfepramone • DCA • DFMDA • DMA • DMMA • EBDB • EDMA • Ethylone • Etolorex • Fenfluramine (Dexfenfluramine) • Flephedrone • IAP • IMP • Lophophine • MBDB • MDA • MDEA • MDHMA • MDMA • MDMPEA • MDOH • MDPEA • Mephedrone • Methedrone • Methylone • MMA • MMDA • MMDMA • MMMA • NAP • Norfenfluramine • 4-TFMA • pBA • pCA • pIA • PMA • PMEA • PMMA • TAP; Piperazines: 2C-B-BZP • 2-BZP • 3-MeOPP • BZP • DCPP • MBZP • mCPP • MDBZP • MeOPP • Mepiprazole • pCPP • pFPP • pTFMPP • TFMPP; Tryptamines: 4-Methyl-αET • 4-Methyl-αMT • 5-CT • 5-MeO-αET • 5-MeO-αMT • 5-MT • αET • αMT • DMT • Tryptamine (itself); Others: Indeloxazine • Tramadol • ViqualineEnzyme inhibitors AGN-2979 • FenclonineNonselective: Benmoxin • Caroxazone • Echinopsidine • Furazolidone • Hydralazine • Indantadol • Iproclozide • Iproniazid • Isocarboxazid • Isoniazid • Linezolid • Mebanazine • Metfendrazine • Nialamide • Octamoxin • Paraxazone • Phenelzine • Pheniprazine • Phenoxypropazine • Pivalylbenzhydrazine • Procarbazine • Safrazine • Tranylcypromine; MAO-A Selective: Amiflamine • Bazinaprine • Befloxatone • Befol • Brofaromine • Cimoxatone • Clorgiline • Esuprone • Harmala alkaloids (Harmine, Harmaline, Tetrahydroharmine, Harman, Norharman, etc) • Methylene Blue • Metralindole • Minaprine • Moclobemide • Pirlindole • Sercloremine • Tetrindole • Toloxatone • TyrimaOthers Ferrous iron (Fe2+) • Magnesium (Mg2+) • Tetrahydrobiopterin • Vitamin B3 (Niacin, Nicotinamide → NADPH) • Vitamin B6 (Pyridoxine, Pyridoxamine, Pyridoxal → Pyridoxal phosphate) • Vitamin B9 (Folic Acid → Tetrahydrofolic acid) • Vitamin C (Ascorbic acid) • Zinc (Zn2+)OthersAntiemetics (A04) 5-HT3 Antagonists Alosetron • Azasetron • Bemesetron • Cilansetron • Clozapine • Dazopride • Dolasetron • Granisetron • Lerisetron • Metoclopramide • Mianserin • Mirtazapine • Olanzapine • Ondansetron • Palonosetron • Ramosetron • Ricasetron • Tropisetron • ZatosetronCB1 Agonists (Cannabinoids) D2/D3 Antagonists H1 Antagonists (Antihistamines) mACh Antagonists (Anticholinergics) NK1 Antagonists Others Drugs for functional gastrointestinal disorders (A03) Drugs for
functional bowel disordersTertiary
amino groupQuaternary ammonium
compoundsBenzilone • Mepenzolate • Pipenzolate • Glycopyrronium • Oxyphenonium • Penthienate • Methantheline • Propantheline • Otilonium bromide • Tridihexethyl • Isopropamide • Hexocyclium • Poldine • Bevonium • Diphemanil • Tiemonium iodide • Prifinium bromide • Timepidium bromide • FenpiveriniumActing on serotonin receptorsOtherFenpiprane • Diisopromine • Chlorbenzoxamine • Pinaverium • Fenoverine • Idanpramine • Proxazole • Alverine • Trepibutone • Isometheptene • Caroverine • Phloroglucinol • Silicones • TrimethyldiphenylpropylamineBelladonna and derivatives
(antimuscarinics)tertiary amines: Atropine • Hyoscyamine
quaternary ammonium compounds: Scopolamine (Butylscopolamine, Methylscopolamine) • Methylatropine • Fentonium • Cimetropium bromidePropulsives Categories:- Drug discovery
- 5-HT3 antagonists
- Antiemetics







Wikimedia Foundation. 2010.