- Discovery and development of cyclooxygenase 2 inhibitors
-
Cyclooxygenases are enzymes that take part in a complex biosynthetic cascade that results in the conversion of polyunsaturated fatty acids to prostaglandins and thromboxane(s).[1] Their main role is to catalyze the transformation of arachidonic acid into the intermediate prostaglandin H2, which is the procursor of a variety of prostanoids with diverse and potent biological actions.[2] Cyclooxygenases have two main isoforms that are called COX-1 and COX-2 (as well as a COX-3). COX-1 is responsible for the synthesis of prostaglandin and thromboxane in many types of cells, including the gastro-intestinal tract and blood platelets. COX-2 plays a major role in prostaglandin biosynthesis in inflammatory cells and in the central nervous system. Prostaglandin synthesis in these sites is a key factor in the development of inflammation and hyperalgesia.[3] COX-2 inhibitors have analgesic and anti-inflammatory activity by blocking the transformation of arachidonic acid into prostaglandin H2 selectively.[4]
Contents
The rise for development of selective COX-2 inhibitors
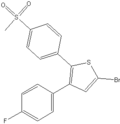
DuP-697 The impetus for development of selective COX-2 inhibitors was the adverse gastrointestinal side-effects of NSAIDs. Soon after the discovery of the mechanism of action of NSAIDs, strong indications emerged for alternative forms of COX, but little supporting evidence was found. COX enzyme proved to be difficult to purify and was not sequenced until 1988.[5] But in 1991 the COX-2 enzyme was cloned and its existence, therefore, confirmed. Before the confirmed existence of COX-2, the Dupont company had developed a compound, DuP-697, that was potent in many anti-inflammatory assays but did not have the ulcerogenic effects of NSAIDs. Once the COX-2 enzyme was identified Dup-697 became the building-block for synthesis of COX-2 inhibitors. Celecoxib and rofecoxib, the first COX-2 inhibitors to reach market, were based on DuP-697.[5][6] It took less than eight years to develop and market the first COX-2 inhibitor, with Celebrex (celecoxib) launched in December 1998 and Vioxx (rofecoxib) launched in May 1999.[7][8]
Development of COX-2 inhibitors
Early studies showed that, when inflammation is induced, the affected organ unexpectedly develops an enormous capacity to generate prostaglandins. It was demonstrated that the increase is due to de novo synthesis of fresh enzyme. In 1991, during the investigation of the expression of early-response genes in fibroblasts transformed with Rous sarcoma virus, a novel mRNA transcript that was similar, but not identical, to the seminal COX enzyme was identified. It was suggested that an isoenzyme of COX had been discovered. Another group discovered a novel cDNA species encoding a protein with similar structure to COX-1 while studying phorbol-ester-induced genes in Swiss 3T3 cells. The same laboratory showed that this gene truly expressed a novel COX enzyme. The two enzymes were renamed COX-1, referring to the original enzyme and COX-2.[5] Building on those results, scientists started focusing on selective COX-2 inhibitors. Enormous effort was spent on the development of NSAIDs between the 1960s and 1980 so there were numerous pharmacophores to test when COX-2 was discovered. Early efforts focused on modification on two lead compounds, DuP-697 and NS-398. These compounds differ greatly from NSAIDs that are arylalkonic acid analogs. Encouraged by the “concept testing” experiments with selective inhibitors, and armed with several solid leads and clear idea of the nature of the binding site, development of this field was rapid.[3] In vitro recombinant enzyme assays provided powerful means for assessing COX selectivity and potency and led to the discovery and clinical development of the first rationally designed COX-2 selective inhibitor, celecoxib. Efforts have been made to convert NSAIDs into selective COX-2 inhibitors such as indometacin by lengthening of the alkylcarboxylic acid side-chain, but none have been marketed.[1]
Structure Activity Relationship (SAR)
DuP-697 was a building-block for synthesis of COX-2 inhibitors and served as the basic chemical model for the coxibs that are the only selective COX-2 inhibitors on the market today. DuP-697 is a diaryl heterocycle with cis-stilbene moiety. Structure activity relationship (SAR) studies for diaryl heterocyclic compounds have indicated that a cis-stilbene moiety and changes in the para-position of one of the aryl rings play an important role in COX-2 selectivity.[1][9] Celecoxib and parecoxib have a sulfonamide substituent (SO2NH2) in para-position on one of the aryl rings while etoricoxib and rofecoxib have a methylsulfone (SO2CH3).[10] The oxidation state on the sulfur is important for selectivity; sulfones and sulfonamides are selective for COX-2 but sulfoxides and sulfides are not. The ring system that is fused in this stilbene system has been extensively manipulated to include every imaginable heterocyclic and carbocyclic skeleton of varying ring sizes. It is known that a SO2NHCOCH3 moiety as in parecoxib, which is a prodrug for valdecoxib, is 105 – 106 more reactive acetylating agent of enzyme serine hydroxyl groups than simple amides.[9] Due to the fact that varying kinetic mechanisms affect potency for COX-1 versus COX-2, relying Potency and selectivity in human whole blood is used by many groups and has been accepted as a standard assessment of COX-2 potency and selectivity.
The relationship between amino acid profile of COX-2 enzyme and inhibition mechanism
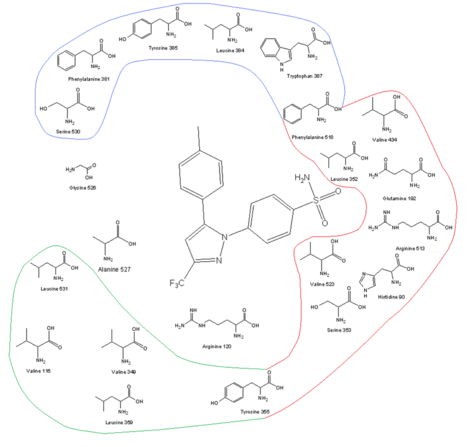
COX-2 receptor site and its amino acid profile along with celecoxib in the binding site One of the keys to developing COX-2 selective drugs is the larger active site of COX-2, which makes it possible to make molecules too large to fit into the COX-1 active site but still able to fit the COX-2. The larger active site of COX-2 is partly due to a polar hydrophilic side-pocket that forms because of substitution of Ile523, His513, and Ile434 in COX-1 by Val523, Arg513, and Val434 in COX-2. Val523 is less bulky than Ile523, which increases the volume of the active site. Substitution of Ile434 for Val434 allows the side-chain of Phe518 to move back and make some extra space. This side-pocket allows for interactions with Arg513, which is a replacement for His513 of COX-1. Arg513 is thought to be a key residue for diaryl heterocycle inhibitors such as the coxibs. The side-chain of Leu384, at the top of the receptor channel, is oriented into the active site of COX-1, but, in COX-2, it is oriented away from the active site and makes more space in the apex of the binding site.[11][12] The bulky sulfonamide group in COX-2 inhibitors such as celecoxib and rofecoxib prevent the molecule from entering the COX-1 channel. For optimal activity and selectivity of the coxibs, a 4-methylsulfonylphenyl attached to an unsaturated (usually) five-membered ring with a vicinal lipophilic group is required (rofecoxib). The SO2CH3 can be replaced by SO2NH2, wherein the lipophilic pocket is occupied by an optionally substituted phenyl ring or a bulky alkoxy substituent (celecoxib). Within the hydrophilic side-pocket of COX-2, the oxygen of the sulfonamide (or sulfone) group interacts with Hist90, Arg513, and Gln192 and forms hydrogen bonds. The substituted phenyl group at the top of the channel interacts with the side-chains of amino acid residues through hydrophobic and electrostatic interactions. Tyr385 makes for some sterical restrictions of this side of the binding site so a small substituent of the phenyl group makes for better binding. Degrees of freedom are also important for the binding. The central ring of the coxibs decides the orientation of the aromatic rings and, therefore, the binding to COX enzyme even though it often has no electrostatic interactions with any of the amino acid residues. The high lipophilicity of the active site does require low polarity of the central scaffold of the coxibs.[12][13]
Mechanism of binding
Studies on the binding mechanism of selective COX-2 inhibitors show that they have two reversible steps with both COX-1 and COX-2, but the selectivity for COX-2 is due to another step that is slow and irreversible and is seen only in the inhibition of COX-2, not COX-1. The irreversible step has been attributed to the presence of the sulfonamide (or sulfone) that fits into the side-pocket of COX-2. This has been studied using SC-58125 (an analogue of celecoxib) and mutated COX-2, wherein the valine 523 residue was replaced by isoleucine 523. The irreversible inhibition did not happen, but reversible inhibition was noticed. A model has been made to explain this three-step mechanism behind the inhibitory effects of selective COX-2 inhibitors. The first step accounts for the contact of the inhibitor with the gate of the hydrophobic channel (called the lobby region). The second step could account for the movement of the inhibitor from the lobby region to the active site of the COX enzyme. The last step probably represents repositioning of the inhibitor at the active site, which leads to strong interactions of the phenylsulfonamide or phenylsulfone group of the inhibitor and the amino acids of the side pocket.[14] It is directly inhibition to postaglanding
Pharmacokinetics of coxibs
The coxibs are widely distributed throughout the body. All of the coxibs achieve sufficient brain concentrations to have a central analgesic effect, and all reduce prostaglandin formation in inflamed joints. All are well absorbed, but peak concentration may differ between the coxibs. The coxibs are highly protein-bound, and the published estimate of half-lives is variable between the coxibs.[15]
Celecoxib
Celecoxib was the first specific inhibitor of COX-2 approved to treat patients with rheumatism and osteoarthritis. A study showed that the absorption rate, when given orally, is moderate, and peak plasma concentration occurs after about 2–4 hours. However, the extent of absorption is not well known. Celecoxib has the affinity to bind extensively to plasma proteins, especially to plasma albumin. It has an apparent volume of distribution (VD) of 455 +/- 166 L in humans and the area under the plasma concentration-time curve (AUC) increases proportionally to increased oral doses, between 100 and 800 mg. Celecoxib is metabolized primarily by CYP2C9 isoenzyme to carboxylic acid and also by non-CYP-dependent glucuronidation to glucuronide metabolites. The metabolites are excreted in urine and feces, with a small proportion of unchanged drug (2%) in the urine. Its elimination half-life is about 11 hours (6–12 hours) in healthy individuals, but racial differences in drug disposition and pharmacokinetic changes in the elderly have been reported. Patients with chronic renal insufficiency appear to have 43% lower plasma concentration compared to healthy individuals, with a 47% increase in apparent clearance, and it can be expected that patients with mild to moderate hepatic impairment have increased steady-state AUC.[16]
Celecoxib Peak [drug] 2–4 hours 
Protein binding 97% Metabolites Carboxylic acid and glucuronide conjugates Half-life [t1/2] 6–12 hours Parecoxib and valdecoxib
Parecoxib sodium is a water-soluble inactive ester amide prodrug of valdecoxib, a novel second-generation COX-2-specific inhibitor and the first such agent to be developed for injectable use. It is rapidly converted by hepatic enzymatic hydrolysis to the active form valdecoxib. The compound then undergoes another conversion, which involves both cytochrome P450-mediated pathway (CYP2C9, CYP3A4) and non-cytochrome P450-mediated pathway, to hydroxylated metabolite and glucuronide metabolite. The hydroxylated metabolite, that also has weak COX-2-specific inhibitory properties, is then further metabolized by non-cytochrome P450 pathway to a glucuronide metabolite. These metabolites are excreted in the urine.[15] After intra-muscular administration of Parecoxib sodium peak plasma concentration is reached within 15 minutes. The plasma concentration decreases rapidly after administration because of a rather short serum half-life, which is about 15–52 minutes. This can be explained by the rapid formation of Valdecoxib. In contrast to the rapid clearance of Parecoxib, plasma concentration of Valdecoxib declines slowly because of a longer half-life.[17] On the other hand, when Valdecoxib is taken orally it is absorbed rapidly (1–2 hours), but presence of food can delay peak serum concentration. It then undergoes the same metabolism that is described above. It is extensively protein-bound (98%), and the plasma half-life is about 7–8 hours. Note that the half-life can be significantly prolonged in the elderly or those with hepatic impairment, and can lead to drug accumulation.[15] The hydroxyl metabolite reaches its highest mean plasma concentration within 3 to 4 hours from administration, but it is considerably lower than of Valdecoxib or about 1/10 of the plasma levels of Valdecoxib.[17]
Parecoxib Peak [drug] Within 15 minutes 
Protein binding N/A Metabolites Valdecoxib, after hepatic enzymatic hydrolysis Half-life [t1/2] 15–52 minutes Valdecoxib Peak [drug] 2–4 hours, delayed by food 
Protein binding 98% Metabolites Hydroxyl derivatives and glucuronide metabolite Half-life [t1/2] 7–8 hours Etoricoxib
Etoricoxib, that is used for patients with chronic arthropathies and musculoskeletal and dental pain, is absorbed moderately when given orally. A study on its pharmacokinetics showed that the plasma peak concentration of etoricoxib occurs after approximately 1 hour. It has shown to be extensively bound to plasma albumin (about 90%), and has an apparent volume of distribution (VD) of 120 L in humans. The area under the plasma concentration-time curve (AUC) increases in proportion to increased dosage (5–120 mg). The elimination half-life is about 20 hours in healthy individuals, and such long half-life enables the choice to have once-daily dosage. Etoricoxib, like the other coxibs, is excreted in urine and feces and also metabolized in likewise manner. CYP3A4 is mostly responsible for biotransformation of etoricoxib to carboxylic acid metabolite, but a non CYP450 metabolism pathway to glucuronide metabolite is also at hand. A very small portion of etoricoxib (<1%) is eliminated unchanged in the urine. Patients with chronic renal insufficiency do not appear to have different plasma concentration curve (AUC) compared to healthy individuals. It has though been reported that patients with moderate hepatic impairment have increased plasma concentration curve (AUC) by approximately 40%. It has been stated that further study is necessary to describe precisely the relevance of pharmacokinetic properties in terms of the clinical benefits and risks of etoricoxib compared to other clinical options.[18][19]
Etoricoxib Peak [drug] 1 hour 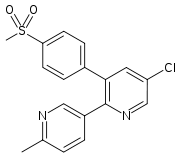
Protein binding 90% Metabolites Carboxylic acid metabolite and glucuronide metabolite Half-life [t1/2] 20 hours Lumiracoxib
Lumiracoxib is unique amongst the coxibs in being a weak acid. It was developed for the treatment of osteoarthritis, rheumatoid arthritis and acute pain. The acidic nature of lumiracoxib allows it to penetrate well into areas of inflammation. It has shown to be rapidly and well absorbed, with peak plasma concentration occurring in about 1–3 hours.[15] A study showed that when a subject was given 400 mg dose, the amount of unchanged drug in the plasma 2.5 hours postdose suggest a modest first pass effect. The terminal half-life in plasma ranged from 5.4 to 8.6 hours (mean =6.5 hours). The half-life in synovial fluid is considerably longer than in plasma, and the concentration in synovial fluid 24 hours after administration would be expected to result in a substantial COX-2 inhibition. This fact can explain why some users may suffice with once-daily dosage despite a short plasma half-life. The major plasma metabolites are 5-carboxy, 4’-hydroxy, and 4’-hydroxy-5-carboxy derivatives. Lumiracoxib is extensively metabolized before it is excreted, and the excretion routes are in the urine or feces. Peak plasma concentrations exceed those necessary to maximally inhibit COX-2, and that is consistent with a longer pharmacodynamic half-life. In vitro lumiracoxib has demonstrated a greater COX-2 selectivity than any of the other coxibs.[20]
Lumiracoxib Peak [drug] 1–3 hours 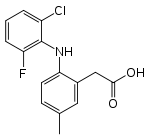
Protein binding 90% Metabolites 5-carboxy, 4’-hydroxy, and 4’-hydroxy-5-carboxy derivatives Half-life [t1/2] 6,5 hours Rofecoxib
Rofecoxib was the second selective COX-2 inhibitor to be marketed, and the first one to be taken off the market.[8] When the pharmacokinetics were studied in healthy human subjects, the peak concentration was achieved in 9 hours with effective half-life of approximately 17 hours . A secondary peak has been observed, which might suggest that the absorption of rofecoxib varies with intestinal motility, hence leading to high variability in time until peak concentration is met. Seventy-one and a half percent of the dose was recovered in urine (less than 1% unmetabolised) and 14,2% was recovered in feces (approximately 1,8% in the bile). Among the metabolites were rofecoxib-3’,4’-dihydrodiol, 4’-hydroxyrofecoxib-O-β-D-glucuronide, 5-hydroxyrofecoxib-O-β-D-glucuronide, 5-hydroxyrofecoxib, rofecoxib-erythro-3,4-dihydrohydroxy acid, rofecoxib-threo-3,4-dihydrohydroxy acid, cis-3,4-dihydrorofecoxib and trans-3,4-dihydrorofecoxib.[21]
Rofecoxib Peak [drug] 9 hours 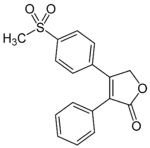
Protein binding N/A Metabolites Major: rofecoxib-threo-3,4-dihydrohydroxy acid and rofecoxib-erythro-3,4-dihydrohydroxy acid Half-life [t1/2] 17 hours Cardiovascular events associated with selective COX-2 inhibitors
Even before the first selective COX-2 inhibitor was marketed, specialists began to suspect that there might be a cardiovascular risk associated with this class of medicines. In the VIGOR study (Vioxx Gastrointestinal Outcomes Research), rofecoxib (Vioxx) was compared to naproxen. After a short time, it became evident that there was a fivefold higher risk of myocardial infarction in the rofecoxib group compared to the group that received naproxen. The authors suggested that the difference was due to the cardioprotective effects of naproxen.[22] The APPROVe (Adenomatous Poly Prevention on Vioxx) study was a multicentre, randomized, placebo-controlled, double blind trial aimed to assess the effect of three-year treatment with rofecoxib on recurrence of neoplastic polyps in individuals with a history of colorectal adenomas.[23][24] In 2000 and 2001, 2587 patients with a history of colorectal adenomas were recruited and followed. The trial was stopped early (2 months before expected completion) on recommendations of its data safety and monitoring board because of concerns about cardiovascular toxicity.[23] When looking at the results of the study, it showed a statistically significant increase in cardiovascular risk when taking rofecoxib compared to placebo[23][24] beginning after 18 months of treatment.[25][23][24] Then on the 30th of September Merck gave out a news release announcing their voluntary worldwide withdrawal of Vioxx.[25] Some studies of other coxibs have also shown increase in the risk of cardiovascular events, while others have not. For instance, the Adenoma Prevention with Celecoxib study (APC) showed a dose-related increase in risk of cardiovascular death, myocardial infarction, stroke, or heart failure when taking celecoxib compared to placebo; and the Successive Celecoxib Efficacy and Safety Study I (SUCCESS-I) showed increased risk of myocardial infarction when taking 100 mg twice a day of celecoxib compared to diclofenac and naproxen; but taking 200 mg twice a day had lower incidence of myocardial infarction compared to diclofenac and naproxen. Nussmeier et al. (2005) showed in a study increase in incidence of cardiovascular events when taking parecoxib and valdecoxib (compared to placebo) after coronary artery bypass surgery.[24]
Possible mechanisms
It has been proposed that COX-2 selectivity could cause imbalance of prostaglandins in the vasculature. If this were the explanation for the increased cardiovascular risk then low-dose aspirin should negate this effect,[24][26] which was not the case in the APPROVe trial.[26] Also, the non-selective COX inhibitors, have also shown increase in cardiovascular events.[27] Another possible explanation was studied by Li H. et al. (2008). They showed that in spontaneously hypertensive rats (SHR) non-selective NSAIDs and the coxibs produce oxidative stress, indicated by enhanced vascular superoxide(O2-) content and elevated peroxide in plasma, which is in tune with enhanced expression of NADPH oxidase, which was noticed with use of diclofenac and naproxen and, to a lesser degree, rofecoxib and celecoxib. Nitrite in plasma was also decreased suggesting a diminished synthesis of vascular nitric oxide (NO). This decrease in NO synthesis did not result from decreased expression of endothelial nitric oxide synthase (eNOS) because expression of eNOS mRNA was not reduced, and even upregulated for some products. The decrease in NO synthesis could, rather, be explained by loss of eNOS function.[27] For eNOS to be normally functional, it needs to form a dimer and to have its cofactor BH4, which is one of the most potent naturally occurring reducing agents. BH4 is sensitive to oxidation by peroxynitrite (ONOO-), which is produced when NO reacts with O2-, so it has been hypothesized that depletion of BH4 can occur with excessive oxidative stress (that can be caused be NSAIDs) and, hence, be the cause of eNOS dysfunction. This disfunction, which is referred to as eNOS uncoupling, causes the production of O2- by eNOS, thereby leading to more oxidative stress produced by eNOS.[28] In a study, both the selective COX-2 inhibitors and the non-selective NSAIDs produced oxidative stress, with greater effects seen with non-selective NSAIDs use. This could fit with the hypothesis concerning the prostacyclin/thromboxane imbalance. That is, although the non-selective NSAIDs produce more oxidative stress, they prevent platelet aggregation, whereas the COX-2 inhibitors reduce prostacyclin production, and, hence, the cardiovascular risk for the non-selective NSAIDs is not higher than for the coxibs.[27] Among other hypotheses are increased blood pressure, decreased production of epi-lipoxins (which have anti-inflammatory effects), and inhibition of vascular remodeling when using selective COX-2 inhibitors.[24]
References
- ^ a b c Marnett, L. J. and A. S. Kalgutkar (1999). "Cyclooxygenase 2 inhibitors: discovery, selectivity and the future." Trends in Pharmacological Sciences 20(11): 465-469. doi:10.1016/S0165-6147(99)01385-1
- ^ Mardini, I. A. and G. A. FitzGerald (2001). "Selective Inhibitors of Cyclooxygenase-2: A Growing Class of Anti-Inflammatory Drugs." Mol. Interv. 1(1): 30-38. [1]
- ^ a b Marnett, L. J. and A. S. Kalgutkar (1998). "Design of selective inhibitors of cyclooxygenase-2 as nonulcerogenic anti-inflammatory agents." Current Opinion in Chemical Biology 2(4): 482-490. doi:10.1016/S1367-5931(98)80124-5
- ^ King, F. D., Ed. (2002). Medicinal chemistry Principles and practice. Cambridge, The royal Society of Chemistry.
- ^ a b c Flower, R. J. (2003). "Case history: The development of COX-2 inhibitors." Nature Reviews Drug Discovery 2(3): 179. [2]
- ^ Dannhardt, G. and W. Kiefer (2001). "Cyclooxygenase inhibitors - current status and future prospects." European Journal of Medicinal Chemistry 36(2): 109-126. doi:10.1016/S0223-5234(01)01197-7
- ^ FDA, Center for Drug Evaluation and Research (2008). "FDA approved drug products - Celebrex." Retrieved 18.10., 2008, from [3]
- ^ a b FDA, Center for Drug Evaluation and Research (2008). "FDA approved drug products - Vioxx." Retrieved 18.10., 2008, from [4]
- ^ a b Zarghi, A., P. N. Rao, et al. (2007). "Design and synthesis of new rofecoxib analogs as selective cyclooxygenase-2 (COX-2) inhibitors: replacement of the methanesulfonyl pharmacophore by a N-acetylsulfonamido bioisostere." Journal of pharmacy & pharmaceutical sciences 10(2): 159-167.[5]
- ^ Mattia, C. and F. Coluzzy (2005). "COX-2 inhibitors: pharmacological data and adverse effects." Minerva anestesiologica 71(7-8): 461-470.[6]
- ^ Llorens, O., J. J. Perez, et al. (1999). "Structural basis of the dynamic mechanism of ligand binding to cyclooxygenase." Bioorganic & Medicinal Chemistry Letters 9(19): 2779-2784. doi:10.1016/S0960-894X(99)00481-3
- ^ a b Michaux, C. and C. Charlier (2004). "Structural Approach for COX-2 Inhibition." Mini Reviews in Medicinal Chemistry 4(6): 603-615.ISSN: 13895575 [7]
- ^ Ermondi, G., G. Caron, et al. (2004). "Docking studies on NSAID/COX-2 isozyme complexes using Contact Statistics analysis." Journal of Computer-Aided Molecular Design 18(11): 683-696. doi:10.1007/s10822-004-6258-1
- ^ Walker, M. C., R. G. Kurumbail, et al. (2001). "A three-step kinetic mechanism for selective inhibition of cyclo-oxygenase-2 by diarylheterocyclic inhibitors." Biochem. J. 357(3): 709-718.[8]
- ^ a b c d Burke, A., E. Smyth, et al. (2005). Ch.26: Analgesic-antipyretic agents; pharmacotherapy of gout. The Pharmacological Basis of THERAPEUTICS. L. L. Brunton, J. S. Lazo and K. L. Parker, McGraw-Hill companies: 679-680 and 702-705.
- ^ Davies, N. M., A. J. McLachlan, et al. (2000). "Clinical Pharmacokinetics and Pharmacodynamics of Celecoxib: A Selective Cyclo-Oxygenase-2 Inhibitor." Clinical Pharmacokinetics 38: 225-242.[9]
- ^ a b Karim, A., A. Laurent, et al. (2001). "A pharmacokinetic study of intramuscular (i.m.) parecoxib sodium in normal subjects." J Clin Pharmacol 41(10): 1111-1119. [10]
- ^ Takemoto, J. K., J. K. Reynolds, et al. (2008). "Clinical Pharmacokinetic and Pharmacodynamic Profile of Etoricoxib." Clinical Pharmacokinetics 47: 703-720. [11]
- ^ Agrawal, N. G. B., A. G. Porras, et al. (2003). "Single- and Multiple-Dose Pharmacokinetics of Etoricoxib, a Selective Inhibitor of Cyclooxygenase-2, in Man." J Clin Pharmacol 43(3): 268-276. doi:10.1177/0091270003251122
- ^ Mangold, J. B., H. Gu, et al. (2004). "Pharmacokinetics and Metabolism of Lumiracoxib in Healthy Male Subjects." Drug Metab Dispos 32(5): 566-571. doi:10.1124/dmd.32.5.566
- ^ Halpin, R. A., L. A. Geer, et al. (2000). "The Absorption, Distribution, Metabolism and Excretion of Rofecoxib, a Potent and Selective Cyclooxygenase-2 Inhibitor, in Rats and Dogs." Drug Metab Dispos 28(10): 1244-1254. [12]
- ^ Jaksch, W., C. Dejaco, et al. (2008). "4 years after withdrawal of rofecoxib: where do we stand today?" Rheumatology International 28(12): 1187-1195. doi:10.1007/s00296-008-0650-4
- ^ a b c d Baron, J. A., R. S. Sandler, et al. (2008). "Cardiovascular events associated with rofecoxib: final analysis of the APPROVe trial." The Lancet In Press, Corrected Proof. [doi:10.1016/S0140-6736(08)61490-7]
- ^ a b c d e f Salinas, G., U. C. Rangasetty, et al. (2007). "The Cycloxygenase 2 (COX-2) Story: It's Time to Explain, Not Inflame." Journal of Cardiovascular Pharmacology and Therapeutics 12(2): 98-111. doi:10.1177/1074248407301172
- ^ a b http://www.merck.com/newsroom/vioxx/pdf/vioxx_press_release_final.pdf
- ^ a b Ferrario, C. M. (2008). "Editorial: On the selective inhibitors of Cyclooxygenase-2: Do we have a last word?" Therapeutic Advances in Cardiovascular Disease 2(2): 75-78.doi:10.1177/1753944708091000
- ^ a b c Li, H., M. Hortmann, et al. (2008). "Cyclooxygenase 2-Selective and Nonselective Nonsteroidal Anti-Inflammatory Drugs Induce Oxidative Stress by Up-Regulating Vascular NADPH Oxidases." J Pharmacol Exp Ther 326(3): 745-753. doi:10.1124/jpet.108.139030
- ^ Forstermann, U. and T. Munzel (2006). "Endothelial Nitric Oxide Synthase in Vascular Disease: From Marvel to Menace." Circulation 113(13): 1708-1714. doi:10.1161/circulationaha.105.602532
Drug design steps in design Case studies of discovery and development of drug classes triptans · cephalosporins · antiandrogens · Bcr-Abl tyrosine kinase inhibitors · nucleoside and nucleotide reverse transcriptase inhibitors · melatonin receptor agonists · proton pump inhibitors · angiotensin receptor blockers · dual serotonin and norepinephrine reuptake inhibitors · cyclooxygenase 2 inhibitors · TRPV1 antagonistsCategories:- Drug discovery
- COX-2 inhibitors








Wikimedia Foundation. 2010.